明膠藥物載體、明膠顆粒栓塞劑及其制備方法和應用與流程
本發明涉及明膠藥物載體、明膠顆粒栓塞劑及其制備方法和應用。
背景技術:
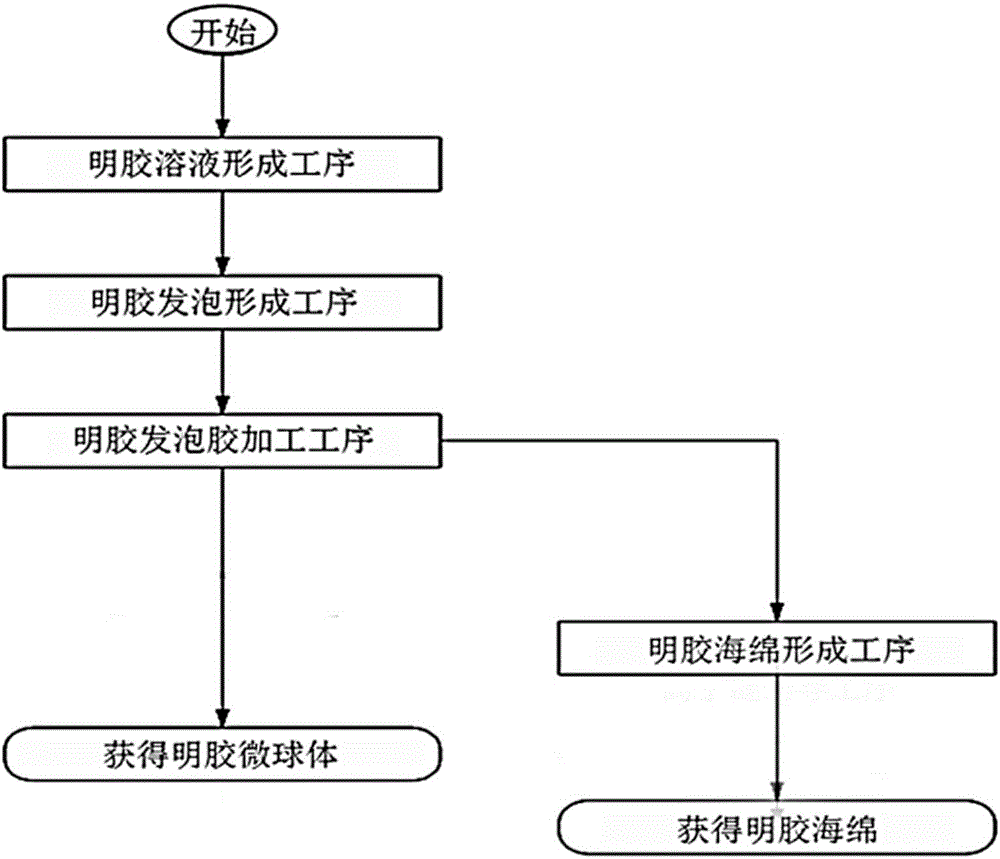
:一般來說,肝癌(Hepatoma)是世界上最常見的癌中的一種,特別是男性中較多見。肝癌的主要誘因是由B型肝炎病毒引起的慢性肝炎和由C型肝炎病毒引起的慢性肝炎,由這些病毒引起的慢性肝炎會導致肝硬化,如果炎癥常時間持續,則極易發生肝癌。此外,酒精性肝疾患或如黃曲霉毒素一樣的毒素引起的肝損傷等慢性肝疾患也都有可能成為肝癌的誘因。肝癌的擴散速度很快,因此早期診斷極其重要。作為治療肝癌的方法有:通過手術去除癌組織的方法;利用肝動脈化療栓塞術(TransarterialChemoembolzation)的方法;和利用經皮(瘤內)無水乙醇注射(PercutaneousEthanolInjection)、高頻熱消融治療(RadiofrequencyThermalAblation)來殺死癌細胞的方法等。但是,如果肝瘤很大,或有大血管侵入,或癌已轉移到了其他臟器時,上述方法就會無效。此外,肝癌治療還有肝切除術、肝移植、局部治療(例如局部高頻熱消融治療、局部乙醇注射術)等方法。但是,我國肝癌細胞患者中約50%以上是在已擴散的病期、或肝硬化已擴散的狀態下診斷出,因此肝動脈化療栓塞術、靶向治療抗癌藥、外部照射放射線治療等非根治治療的作用也是很重要的。在非根治肝癌治療的肝動脈化療栓塞術中,有一種新的肝動脈治療術,即利用藥物緩釋微球(drug-elutingmicrobead)的肝癌治療方法。在該方法中,藥物緩釋微球為100~700μm大小的微球與抗癌藥物結合的栓塞物,并利用肝動脈來治療肝癌。現有的藥物緩釋微球是由聚乙烯醇(polyvinylalcohol)制作的微珠(Bead)等形態的永久性栓塞物,對肝血管的損傷大,肝血管被永久性地堵塞,當對復發率高的肝癌再次用化療栓塞術進行治療時,其難度加重。因而,開發一種不會對肝功能造成障礙特別是對肝癌有效的藥物緩釋微球變得很緊迫。本申請人于2015年7月29日申請并于同年12月1日注冊的“用于肝動脈化療栓塞術的含有磷酸鹽緩沖鹽水或磷酸鹽緩沖液的明膠海綿及其制造方法”中公開了以下內容:在4~37℃的磷酸鹽緩沖液(PBS)或磷酸緩沖液(PhosphateBuffer)中浸漬明膠海綿3~12小時的工序;將沉淀有磷酸鹽緩沖液(PBS)或磷酸緩沖液(PhosphateBuffer)的明膠海綿在-10℃~-35℃的溫度條件下冷凍干燥30分鐘到16小時的工序;對上述冷凍干燥后的明膠海綿進行粉碎的工序;把上述被粉碎的明膠海綿分離成10~100μm、100~300μm、300~500μm、500~700μm的大小后,放入消毒后的小藥瓶或放入裝有磷酸鹽緩沖鹽水或磷酸鹽緩沖液中的任一個的包裝容器中的狀態下保管,之后投入生理鹽水、蒸餾水和抗癌藥,所投入的生理鹽水、蒸餾水和抗癌藥相互鍵合形成離子鍵,以作為一種肝癌治療藥。上述公開的技術方案具有工序比較簡單、制備比較容易的優點,但是上述方案中使用的初始明膠海綿為市售產品,在制造過程中進行過冷凍干燥,而在將已經冷凍干燥完畢的明膠海綿浸漬到磷酸鹽緩沖液或者磷酸緩沖液之后進行再制冷干燥的過程中,海綿的多孔尺寸無法避免地會被破壞,因此存在很難控制其形態的問題和在利用粉碎機粉碎明膠海綿的工藝中因出現栓塞劑顆粒的形態不均勻或者表面尺寸似被剪切般不均勻問題,存在很難控制產品形態的問題。而且,所公開的工藝如果無法適當地調節凍結干燥時的水分含量,磷酸緩沖液會形成為粉末狀,會引發與明膠海綿結塊兒的缺點,因此也需要改善其附加工藝。技術實現要素:本發明所解決的技術問題在于克服現有的明膠海綿存在再制冷干燥會產生多孔尺寸破壞乃至粉碎時引起的栓塞劑顆粒形態不規則的缺陷,提供了明膠藥物載體、明膠顆粒栓塞劑及其制備方法和應用。本發明通過將交聯化的明膠發泡膠添加至磷酸鹽緩沖鹽水(PBS)、磷酸緩沖液(PhosphateBuffer)、Tris(三羥甲基氨基甲烷)緩沖液或者glycine(甘氨酸)緩沖液中并保持pH值在7以上,有效地改善了因再制冷干燥引發的問題。本發明的明膠顆粒栓塞劑通過將含有磷酸鹽緩沖鹽水、磷酸緩沖液、Tris緩沖液或者glycine緩沖液的明膠藥物載體與抗癌劑結合并將此作為栓塞物質,利用肝動脈對肝癌進行非根治性治療。本發明是通過下述技術方案解決上述技術問題的。本發明提供了一種明膠藥物載體的制備方法,其包括如下步驟:在交聯結構狀態的明膠發泡膠里添加溶液A,并使氫離子濃度保持在從中性至堿性的pH范圍內,然后對其進行干燥,即可;所述溶液A為磷酸鹽緩沖鹽水、磷酸緩沖液、Tris緩沖液或者glycine緩沖液,其中,所述交聯結構狀態的明膠發泡膠通過下述方法制備:在明膠水溶液中添加醛類固化劑,并進行攪拌使明膠溶液固化,或者將明膠水溶液加入到穩定劑中攪拌混勻后,再添加醛類固化劑固化,之后再用丙酮或蒸餾水清洗去除殘留的醛類固化劑,即得交聯結構狀態的明膠發泡膠。本發明中,本領域技術人員根據本領域常識和本發明所述交聯結構狀態的明膠發泡膠的制備方法,可知曉本發明所述交聯結構狀態的明膠發泡膠呈現水溶液狀態,并且知曉所述交聯結構狀態的明膠發泡膠在制備過程中,明膠水溶液和醛類固化劑進行交聯反應后在溶液內形成氣孔,具體狀態圖可參見附圖1的照片。本發明中,所述交聯結構狀態的明膠發泡膠在添加磷酸鹽緩沖鹽水、磷酸緩沖液、Tris緩沖液或者glycine緩沖液前,還可先進行如下步驟:將所述交聯結構狀態的明膠發泡膠進行冷凍,得到凍結狀態的明膠發泡膠,之后再以蒸餾水浸漬或者漂洗的方式進行清洗并脫水,以清洗除去明膠發泡膠上殘留的甲醛。其中,所述冷凍的溫度較佳地為-35~-45℃,所述冷凍的時間較佳地為12小時以上,例如12~16h。其中,所述凍結狀態的明膠發泡膠還可先進行粉碎,再用蒸餾水浸漬或者漂洗的方式進行清洗并脫水。本領域技術人員均知曉將本發明呈現水溶液狀態的所述交聯結構狀態的明膠發泡膠冷凍后可得到堅硬如石頭般的明膠發泡膠。本發明中,所述用丙酮或蒸餾水清洗去除殘留的醛類固化劑可通過用丙酮或蒸餾水反復清洗、離心分離實現,所述離心分離一般利用離心分離機進行,所述清洗的次數較佳地為5次。本發明中,所述磷酸鹽緩沖鹽水、磷酸緩沖液、Tris緩沖液或者glycine緩沖液起到保持pH值的功能,以用于避免pH值發生急劇變化。較佳地,所述溶液A為磷酸鹽緩沖鹽水或者磷酸緩沖液。本發明中,所述磷酸鹽緩沖鹽水為本領域常規使用的磷酸鹽緩沖鹽水,其組分一般包括磷酸二氫鈉或磷酸二氫鉀、磷酸氫二鈉或磷酸氫二鉀以及氯化鈉和/或氯化鉀。所述磷酸鹽緩沖鹽水可按照本領域常規方法配制。所述磷酸緩沖液為本領域常規使用的磷酸緩沖液,其組分一般包括磷酸二氫鈉或磷酸二氫鉀以及磷酸氫二鈉或磷酸氫二鉀。所述磷酸緩沖液可按照本領域常規方法配制。所述Tris緩沖液和glycine緩沖液均為本領域常規使用的Tris緩沖液和glycine緩沖液,皆市售可得。本發明中,使用的磷酸鹽緩沖鹽水、磷酸緩沖液、Tris緩沖液或glycine緩沖液的pH和濃度本領域技術人員可根據實際需要結合本領域常識進行選擇。較佳地,所述磷酸鹽緩沖鹽水的濃度為12.5mM和25mM時,pH為8時最佳;所述磷酸緩沖液的濃度為12.5mM和25mM時,pH為8時最佳;所述Tris緩沖液的濃度為任意濃度時,pH為9時最佳,而濃度為12.5mM和25mM時,pH為8時最佳;所述glycine緩沖液的濃度為25mM時,pH為9時最佳。本發明中,所述干燥的方式為本領域常規操作的方式,例如除濕干燥、氮氣干燥、冷風干燥、真空干燥、自然干燥、冷凍干燥和噴霧干燥中的任一方式。所述干燥為冷凍干燥時,所述冷凍干燥的溫度和時間可按照本領域常規選擇,所述冷凍干燥的溫度一般為-35~-45℃,所述冷凍的時間一般為14小時以上,例如14~16h。較佳地,所述冷凍干燥為干燥至水分含量為10~20%,更佳地,所述冷凍干燥為干燥至水分含量為10~15%。在本發明一較佳實施例中,所述明膠藥物載體的制備方法包括如下步驟:(1)明膠溶液形成工序:把明膠放入100mL的水中,浸泡30分鐘并加熱攪拌至明膠溶解,然后再降溫至40℃,即得濃度為1~6%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:添加0.1~2mg/mL濃度的甲醛,并以500~1000rpm、1200~1400rpm或1400~1500rpm的速度進行攪拌,使其固化,以此形成明膠發泡膠;(3)離心分離工藝:利用丙酮或者蒸餾水清洗所獲得的明膠發泡膠,然后離心分離;(4)明膠發泡膠加工工序:將離心分離的明膠發泡膠添加至磷酸鹽緩沖鹽水或者磷酸緩沖液中,使其保持pH值7以上,并清洗5次之后進行離心脫水;(5)明膠海綿形成工序:對明膠發泡膠進行冷凍干燥,即得。其中,步驟(1)中,所述明膠溶液的濃度較佳地為4~5%(w/v),所述百分比為明膠溶液中的明膠質量占明膠溶液體積的比值,單位為g/mL。其中,步驟(2)中,所述甲醛的濃度較佳地為0.1~0.5mg/mL,更佳地為0.1~0.3mg/mL。其中,步驟(2)中,所述攪拌的時間可根據本領域常規選擇。較佳地,所述攪拌的速度為1400~1500rpm,所述攪拌的時間為15~30秒。其中,步驟(3)中,所述離心分離可采用本領域常規設備進行,例如離心分離機。其中,步驟(5)中,所述冷凍干燥可采用本領域常規設備進行,一般采用冷凍干燥機。在本發明另一較佳實施例中,所述明膠藥物載體的制備方法包括如下步驟:(1)明膠溶液形成工序:把明膠溶解于30~70℃的水中,并通過對其過濾獲得15~60%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:把明膠溶液放入質量比為1~2%的穩定劑span80當中,并使得水與油的體積比為1/1~1/10,以200~800r/min攪拌15秒,然后在10℃溫度下,添加質量比為2~50%的醛類交聯劑化合物,并凝固1~2h之后,通過清洗或者離心分離的方法來清除交聯劑,即得明膠發泡膠;(3)明膠發泡膠加工工序:把明膠發泡膠添加至磷酸鹽緩沖鹽水或者磷酸緩沖液,并保持pH值為7以上,清洗5次以后進行離心脫水;(4)明膠微球體形成工序:將上述明膠發泡膠進行溶解,并進行噴霧干燥,即得。其中,步驟(1)中,所述水的溫度較佳地為55~65℃。其中,步驟(1)中,所述明膠溶液的濃度較佳地為25~30%(w/v),所述百分比為明膠溶液中的明膠質量占明膠溶液體積的比值,單位為g/mL。其中,步驟(2)中,所述穩定劑span80的質量比較佳地為2%,所述質量比的數值與用質量體積比g/mL折算的數值基本一致。其中,步驟(2)中,所述水與油的體積比較佳地為1/1。其中,步驟(2)中,所述醛類交聯劑化合物的質量比較佳地為6~8%,所述質量比的數值與用質量體積比g/mL折算的數值基本一致。本發明還提供了一種由上述制備方法制得的明膠藥物載體。本發明還提供了一種明膠顆粒栓塞劑的制備方法,所述制備方法包括如下步驟:將上述明膠藥物載體粉碎后,以包裝容器的狀態進行保管;并投入肝癌治療藥,使其與肝癌治療藥形成離子鍵。其中,所述粉碎可按照本領域常規操作進行,所述粉粹后較佳地還進行篩分,所述篩分較佳地按照50~100μm、100~300μm、300~500μm、500~700μm、500~700μm、700~1000μm、1000~1500μm、1500μm~2000μm的粒徑進行篩分。其中,所述肝癌治療藥劑為本領域常規的肝癌治療藥劑。本領域技術人員均知曉在投入所述肝癌治療藥劑的同時一般還一并投入生理鹽水、蒸餾水或無菌水稀釋。本發明還提供了一種由上述制備方法制得的明膠顆粒栓塞劑。本發明還提供了一種上述明膠顆粒栓塞劑在肝動脈化療栓塞術中的應用。在不違背本領域常識的基礎上,上述各優選條件,可任意組合,即得本發明各較佳實例。本發明所用試劑和原料均市售可得。本發明的積極進步效果在于:(1)本發明通過將交聯化的明膠發泡膠利用丙酮或蒸餾水清洗后,添加至磷酸鹽緩沖鹽水、磷酸緩沖液、Tris緩沖液或者glycine緩沖液并保持pH值在7以上,有效地改善了因再制冷干燥引發的多孔尺寸的破壞及進行粉碎時產生的栓塞劑顆粒形態出現不規則等問題,這將更加有益于使用于肝動脈化療栓塞術中。(2)本發明通過將肝癌治療藥劑結合到明膠海綿或者明膠微球體中,并將其作為栓塞物質,能夠對肝癌進行非根治性治療,并且具有當復發率較高的肝癌患者需要再次進行手術時,還可以進行利用藥物緩釋微球的化療栓塞術的效果。(3)本發明提供的一種明膠藥物載體——明膠微球體是一種在體內分解的微球,具體為在肝癌治療中注射到肝時經過規定時間后在體內分解的微球,在這期間遮斷對血管型的惡性腫瘤供給的血液,藥物緩釋微球局部地分解并持續地將藥物遞送(DrugDelivery)到腫瘤,從而減少由抗癌藥流入全身血流引起的副作用,并增加腫瘤反應。附圖說明圖1為本發明交聯結構狀態的明膠發泡膠的溶液狀態圖。圖2為本發明明膠藥物載體的制備方法的順序流程示意圖。圖3為本發明交聯結構狀態的明膠發泡膠浸漬在磷酸鹽緩沖鹽水中的狀態的立體示意圖。圖4為本發明交聯結構狀態的發泡膠浸漬在磷酸鹽緩沖液中的狀態的立體示意圖。圖5為本發明的明膠海綿在粉碎成粉末的狀態下放入小藥瓶中包裝處理后的狀態的立體示意圖。圖6為本發明被保管在小藥瓶中的明膠海綿中結合生理鹽水或蒸餾水以及抗癌藥的狀態的立體示意圖。圖7為利用本發明實驗例在420nm波長下檢測的各個濃度的阿雷素吸光度的檢測結果圖。圖8為利用本發明實驗例在450nm波長下檢測的各個濃度的阿雷素吸光度的檢測結果圖。圖9為利用本發明實驗例在510nm波長下檢測的各個濃度的阿雷素吸光度的檢測結果圖。圖10為根據本發明實驗例中水化溶液的種類的明膠粒子的阿雷素吸附能力的吸光度檢測結果圖。圖11為根據本發明實驗例中水化溶液的種類的明膠粒子的阿雷素吸附能力的阿雷素濃度檢測結果圖。圖12為本發明實驗例中從生理鹽水洗脫出來的阿雷素。圖13為對照組明膠海綿的數碼照片(a)和不同放大倍率下的SEM照片(b、c)。圖14為對照組明膠微球體的SEM照片。圖15為本發明明膠海綿的SEM照片。圖16為本發明明膠微球體的顯微鏡照片和SEM照片。附圖標記說明如下:10明膠海綿20磷酸鹽緩沖鹽水30磷酸鹽緩沖液具體實施方式下面通過實施例的方式進一步說明本發明,但并不因此將本發明限制在所述的實施例范圍之中。下列實施例中未注明具體條件的實驗方法,按照常規方法和條件,或按照商品說明書選擇。下述說明書附圖中,與說明無關的部分進行了省略,以更清楚地說明本發明,對于相似的部分,標記相似的附圖標記。實施例1一種明膠藥物載體,其制備流程圖如圖2所示,具體制備方法如下:(1)明膠溶液形成工序:把明膠放入100mL的水中,浸泡30分鐘并加熱攪拌至明膠溶解,然后再降溫至40℃,以獲得濃度為4%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:向所得明膠溶液中添加0.5mg/mL濃度的甲醛,并以1400~1500rpm的速度攪拌15~30秒,使其明膠溶液固化,以此形成交聯結構狀態的明膠發泡膠;(3)離心分離工藝:利用丙酮清洗所獲得的明膠發泡膠,然后利用離心分離機進行離心分離;(4)明膠發泡膠加工工序:將離心分離后的明膠發泡膠添加至pH為8,濃度為12.5mM的磷酸鹽緩沖鹽水中,并使其保持pH值7以上,且清洗5次之后進行離心脫水;其中明膠發泡膠浸漬在磷酸鹽緩沖鹽水中的狀態的立體圖可參見圖3;(5)明膠海綿形成工序:利用冷凍干燥機對上述明膠發泡膠進行干燥,使水分達到10~15%,以此形成明膠海綿。所得明膠海綿的外形和多孔尺寸穩定,孔隙大小均一,孔隙大小和厚度處于最佳狀態。一種明膠顆粒栓塞劑,其由包括如下步驟的方法制備:對上述所得的明膠海綿進行粉碎,粉碎后按照50~100μm、100~300μm、300~500μm、500~700μm粒徑進行篩分,并將篩分選別后的顆粒以包裝容器的狀態進行保管,具體可參見圖5,然后利用生理鹽水稀釋后投入到肝癌治療藥劑中,以此誘導離子鍵耦合,具體可參見圖6;其中,完成粉碎及選別工序的明膠海綿被保管至裝有pH為8,濃度為12.5mM的磷酸鹽緩沖鹽水的包裝容器中保管,然后投入生理鹽水和抗癌藥,使其相互誘導離子耦合,以此作為一種肝癌治療藥使用。實施例2一種明膠藥物載體,其制備流程圖如圖2所示,具體制備方法如下:(1)明膠溶液形成工序:把明膠放入100mL的水中,浸泡30分鐘并加熱攪拌至明膠溶解,然后再降溫至40℃,以獲得濃度為5%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:向所得明膠溶液中添加0.3mg/mL濃度的甲醛,并以1400~1500rpm的速度攪拌15~30秒,使其明膠溶液固化,以此形成交聯結構狀態的明膠發泡膠;(3)離心分離工藝:利用蒸餾水清洗所獲得的明膠發泡膠,然后利用離心分離機進行離心分離;(4)明膠發泡膠加工工序:將離心分離后的明膠發泡膠添加至pH為8,濃度為25mM的磷酸緩沖液中,并使其保持pH值7以上,且清洗5次之后進行離心脫水;其中明膠發泡膠浸漬在磷酸緩沖液中的狀態的立體圖可參見圖4;(5)明膠海綿形成工序:利用冷凍干燥機對上述明膠發泡膠進行干燥,使水分達到10~15%,以此形成明膠海綿。所得明膠海綿的外形和多孔尺寸穩定,孔隙大小均一,孔隙大小和厚度處于最佳狀態。一種明膠顆粒栓塞劑,其由包括如下步驟的方法制備:將上述所得的明膠海綿進行粉碎,粉碎后按照50~100μm、100~300μm、300~500μm、500~700μm粒徑進行篩分,并將篩分選別后的顆粒以包裝容器的狀態進行保管,具體可參見圖5,然后利用蒸餾水稀釋后投入到肝癌治療藥劑中,以此誘導離子鍵耦合,具體可參見圖6;其中,完成粉碎及選別工序的明膠海綿被保管至裝有pH為8,濃度為25mM的磷酸緩沖液的包裝容器中保管,然后投入蒸餾水和抗癌藥,使其相互誘導離子耦合,以此作為一種肝癌治療藥使用。實施例3一種明膠藥物載體,其制備流程圖如圖2所示,具體制備方法如下:(1)明膠溶液形成工序:把明膠放入100mL的水中,浸泡30分鐘并加熱攪拌至明膠溶解,然后再降溫至40℃,以獲得濃度為1%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:向所得明膠溶液中添加0.1mg/mL濃度的甲醛,并以500~1000rpm的速度進行攪拌,使其明膠溶液固化,以此形成交聯結構狀態的明膠發泡膠;(3)離心分離工藝:利用丙酮清洗所獲得的明膠發泡膠,然后利用離心分離機進行離心分離;(4)明膠發泡膠加工工序:將離心分離后的明膠發泡膠添加至pH為8,濃度為12.5mM的Tris緩沖液中,并使其保持pH值7以上,且清洗5次之后進行離心脫水;(5)明膠海綿形成工序:利用冷凍干燥機對上述明膠發泡膠進行干燥,使水分達到15~20%,以此形成明膠海綿。所得明膠海綿的外形和多孔尺寸較為穩定,孔隙大小不均一。一種明膠顆粒栓塞劑,其由包括如下步驟的方法制備:將上述所得的明膠海綿進行粉碎,粉碎后按照50~100μm、100~300μm、300~500μm、500~700μm粒徑進行篩分,并將篩分選別后的顆粒以包裝容器的狀態進行保管,具體可參見圖5,然后利用蒸餾水稀釋后投入到肝癌治療藥劑中,以此誘導離子鍵耦合,具體可參見圖6;其中,完成粉碎及選別工序的明膠海綿被保管至消毒完畢的小藥瓶中保管,然后投入蒸餾水和抗癌藥,使其相互誘導離子耦合,以此作為一種肝癌治療藥使用。實施例4一種明膠藥物載體,其制備流程圖如圖2所示,具體制備方法如下:(1)明膠溶液形成工序:把明膠放入100mL的水中,浸泡30分鐘并加熱攪拌至明膠溶解,然后再降溫至40℃,以獲得濃度為6%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:向所得明膠溶液中添加0.1mg/mL濃度的甲醛,并以1200~1400rpm的速度進行攪拌,使其明膠溶液固化,以此形成交聯結構狀態的明膠發泡膠;(3)離心分離工藝:利用丙酮清洗所獲得的明膠發泡膠,然后利用離心分離機進行離心分離;(4)明膠發泡膠加工工序:將離心分離后的明膠發泡膠添加至pH為9,濃度為25mM的glycine緩沖液中,并使其保持pH值7以上,且清洗5次之后進行離心脫水;(5)明膠海綿形成工序:利用冷凍干燥機對上述明膠發泡膠進行干燥,使水分達到10~15%,以此形成明膠海綿。所得明膠海綿的外形和多孔尺寸較為穩定,孔隙大小和厚度處于較佳狀態。一種明膠顆粒栓塞劑,其由包括如下步驟的方法制備:將上述所得的明膠海綿進行粉碎,粉碎后按照50~100μm、100~300μm、300~500μm、500~700μm粒徑進行篩分,并將篩分選別后的顆粒以包裝容器的狀態進行保管,具體可參見圖5,然后利用蒸餾水稀釋后投入到肝癌治療藥劑中,以此誘導離子鍵耦合,具體可參見圖6;其中,完成粉碎及選別工序的明膠海綿被保管至裝有pH為8,濃度為12.5mM的磷酸鹽緩沖鹽水的包裝容器中保管,然后投入蒸餾水和抗癌藥,使其相互誘導離子耦合,以此作為一種肝癌治療藥使用。實施例5一種明膠藥物載體,其制備流程圖如圖2所示,具體制備方法如下:(1)明膠溶液形成工序:把一定量的明膠溶解于55~65℃的水中,并通過對其過濾獲得濃度為25~30%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:把明膠溶液放入質量比為2%的穩定劑span80當中,并使得水與油的體積比為1/1,以500r/min攪拌15秒,然后在10℃溫度下,添加質量比為6~8%的醛類交聯劑化合物,并凝固1~2h之后,通過清洗或者離心分離的方法來清除交聯劑,即得交聯結構狀態的明膠發泡膠;(3)明膠發泡膠加工工序:把明膠發泡膠添加至pH為8,濃度為25mM的磷酸鹽緩沖鹽水中,并保持pH值為7以上,清洗5次以后進行離心脫水;(4)明膠微球體形成工序:溶解上述明膠發泡膠,利用噴霧干燥機進行噴霧干燥,以此形成明膠微球體。一種明膠顆粒栓塞劑,其由包括如下步驟的方法制備:將上述所得的明膠微球體按照50~100μm、100~300μm、300~500μm、500~700μm、700~1000μm、1000~1500μm的大小進行篩選;篩選后的明膠微球體以包裝容器的狀態進行保管,然后利用生理鹽水稀釋后注入肝癌治療藥劑,以此誘導鍵合離子鍵;其中,完成粉碎及篩選工序的明膠微球體被保管至裝有pH為8,濃度為25mM的磷酸鹽緩沖鹽水的包裝容器中保管,然后投入生理鹽水和抗癌藥,使其相互鍵合形成離子鍵,以此作為一種肝癌治療藥使用。通過上述實施例5獲得的明膠微球體作為微小型球形栓塞劑,已確認到其內部為海綿狀,不僅吸水力優秀,而且尺寸均一,最重要的是確認到其具有一定彈性及膨脹力。實施例6一種明膠藥物載體,其制備流程圖如圖2所示,具體制備方法如下:(1)明膠溶液形成工序:把一定量的明膠溶解于30~40℃的水中,并通過對其過濾獲得濃度為15~25%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:把明膠溶液放入質量比為2%的穩定劑span80當中,并使得水與油的體積比為1/2,以500r/min攪拌15秒,然后在10℃溫度下,添加質量比為2~4%的醛類交聯劑化合物,并凝固1~2h之后,通過清洗或者離心分離的方法來清除交聯劑,即得交聯結構狀態的明膠發泡膠;(3)明膠發泡膠加工工序:把明膠發泡膠添加至pH為8,濃度為12.5mM的磷酸鹽緩沖鹽水中,并保持pH值為7以上,清洗5次以后進行離心脫水;(4)明膠微球體形成工序:溶解上述明膠發泡膠,利用噴霧干燥機進行噴霧干燥,以此形成明膠微球體。一種明膠顆粒栓塞劑,其由包括如下步驟的方法制備:將上述所得的明膠微球體按照50~100μm、100~300μm、300~500μm、500~700μm、700~1000μm、1000~1500μm的大小進行篩選;篩選后的明膠微球體以包裝容器的狀態進行保管,然后利用生理鹽水稀釋后注入肝癌治療藥劑,以此誘導鍵合離子鍵;其中,完成粉碎及篩選工序的明膠微球體被保管至裝有pH為8,濃度為12.5mM的磷酸鹽緩沖鹽水的包裝容器中保管,然后投入生理鹽水和抗癌藥,使其相互鍵合形成離子鍵,以此作為一種肝癌治療藥使用。通過上述實施例6獲得的明膠微球體作為微小型球形栓塞劑,已確認到其內部為海綿狀,不僅吸水力優秀,而且尺寸均一,最重要的是確認到其具有一定彈性及膨脹力。實施例7一種明膠藥物載體,其制備流程圖如圖2所示,具體制備方法如下:(1)明膠溶液形成工序:把一定量的明膠溶解于60~70℃的水中,并通過對其過濾獲得濃度為50~60%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:把明膠溶液放入質量比為1%的穩定劑span80當中,并使得水與油的體積比為1/10,以500r/min攪拌15秒,然后在10℃溫度下,添加質量比為45~50%的醛類交聯劑化合物,并凝固1~2h之后,通過清洗或者離心分離的方法來清除交聯劑,即得交聯結構狀態的明膠發泡膠;(3)明膠發泡膠加工工序:把明膠發泡膠添加至pH為8,濃度為12.5mM的磷酸緩沖液中,并保持pH值為7以上,清洗5次以后進行離心脫水;(4)明膠微球體形成工序:溶解上述明膠發泡膠,利用噴霧干燥機進行噴霧干燥,以此形成明膠微球體。一種明膠顆粒栓塞劑,其由包括如下步驟的方法制備:將上述所得的明膠微球體按照50~100μm、100~300μm、300~500μm、500~700μm、700~1000μm、1000~1500μm的大小進行篩選;篩選后的明膠微球體以包裝容器的狀態進行保管,然后利用蒸餾水稀釋后注入肝癌治療藥劑,以此誘導鍵合離子鍵;其中,完成粉碎及篩選工序的明膠微球體被保管至裝有pH為8,濃度為12.5mM的磷酸緩沖液的包裝容器中保管,然后投入蒸餾水和抗癌藥,使其相互鍵合形成離子鍵,以此作為一種肝癌治療藥使用。通過上述實施例7獲得的明膠微球體作為微小型球形栓塞劑,已確認到其內部為海綿狀,不僅吸水力優秀,而且尺寸均一,最重要的是確認到其具有一定彈性及膨脹力。實施例8一種明膠藥物載體,其制備方法包括如下步驟:(1)明膠溶液形成工序:把明膠放入100mL的水中,浸泡30分鐘并加熱攪拌至明膠溶解,然后再降溫至40℃,以獲得濃度為4%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:向所得明膠溶液中添加0.5mg/mL濃度的甲醛,并以1400~1500rpm的速度攪拌15~30秒,使其明膠溶液固化,以此形成明膠發泡膠,將所得明膠發泡膠在-35~-45℃下冷凍12~16h,得到凍結狀態的明膠發泡膠;(3)離心分離工藝:利用蒸餾水浸漬清洗所獲得的明膠發泡膠,然后利用離心分離機進行離心分離脫水;(4)明膠發泡膠加工工序:將離心分離后的明膠發泡膠添加至pH為8,濃度為25mM的磷酸鹽緩沖鹽水中,并使其保持pH值7以上,且清洗5次之后進行離心脫水;(5)明膠海綿形成工序:利用冷凍干燥機對上述明膠發泡膠在-35~-45℃下冷凍干燥14~16h,以此形成明膠海綿。所得明膠海綿的外形、多孔尺寸穩定性、孔隙大小和厚度與實施例1相當。一種明膠顆粒栓塞劑,其由包括如下步驟的方法制備:將上述所得的明膠海綿按照50~100μm、100~300μm、300~500μm、500~700μm、700~1000μm、1000~1500μm、1500~2000μm的大小進行篩選;篩選后的明膠海綿以包裝容器的狀態進行保管,然后利用蒸餾水稀釋后注入肝癌治療藥劑,以此誘導鍵合離子鍵;其中,完成粉碎及篩選工序的明膠海綿被保管至裝有pH為8,濃度為25mM的磷酸鹽緩沖鹽水的包裝容器中保管,然后投入蒸餾水和抗癌藥,使其相互鍵合形成離子鍵,以此作為一種肝癌治療藥使用。實施例9一種明膠藥物載體,其制備方法包括如下步驟:(1)明膠溶液形成工序:把明膠放入100mL的水中,浸泡30分鐘并加熱攪拌至明膠溶解,然后再降溫至40℃,以獲得濃度為4%(w/v)的明膠溶液;(2)明膠發泡膠形成工序:向所得明膠溶液中添加0.5mg/mL濃度的甲醛,并以1400~1500rpm的速度攪拌15~30秒,使其明膠溶液固化,以此形成明膠發泡膠,將所得明膠發泡膠在-35~-45℃下冷凍12~16h,得到凍結狀態的明膠發泡膠;(3)離心分離工藝:利用蒸餾水漂洗所獲得的明膠發泡膠,然后利用離心分離機進行離心分離脫水;(4)明膠發泡膠加工工序:將離心分離后的明膠發泡膠添加至pH為8,濃度為12.5mM的磷酸鹽緩沖鹽水中,并使其保持pH值7以上,且清洗5次之后進行離心脫水;(5)明膠海綿形成工序:利用冷凍干燥機對上述明膠發泡膠在-35~-45℃下冷凍干燥14~16h,以此形成明膠海綿。所得明膠海綿的外形、多孔尺寸穩定性、孔隙大小和厚度與實施例1相當。一種明膠顆粒栓塞劑,其由包括如下步驟的方法制備:將上述所得的明膠海綿按照50~100μm、100~300μm、300~500μm、500~700μm、700~1000μm、1000~1500μm、1500~2000μm的大小進行篩選;篩選后的明膠海綿以包裝容器的狀態進行保管,然后利用蒸餾水稀釋后注入肝癌治療藥劑,以此誘導鍵合離子鍵;其中,完成粉碎及篩選工序的明膠海綿被保管至裝有pH為8,濃度為12.5mM的磷酸鹽緩沖鹽水的包裝容器中保管,然后投入蒸餾水和抗癌藥,使其相互鍵合形成離子鍵,以此作為一種肝癌治療藥使用。效果實施例1本效果實施例所使用的靜脈注射用阿雷素是日東制藥公司的ADRIAMYCIN-RDF(10mg小藥瓶)。通過把藥劑溶于10mL的無菌蒸餾水的方法制作了1mg/mL的stocksolution,并將其保管于4℃環境下,應用于實驗當中。利用水溶液狀態的阿雷素呈紅色的特性,使用分光光度計來替代精度管理較為復雜的HPLC,確認是否能夠簡單地對阿雷素進行定量。為了在吸光度檢測值和阿雷素濃度之間,具有比例相關性的光源波長,利用多種波長檢測了阿雷素溶液的吸光度。首先,通過把stocksolution稀釋到蒸餾水當中,準備濃度各為20ug/mL、40ug/mL、60ug/mL、80ug/mL、100ug/mL的阿雷素之后,在420nm、450nm及510nm的波長下檢測了各自的吸光度值。檢測結果見下表1和圖6~8。在三種波長當中,都觀察了與各個濃度的阿雷素的吸光度檢測值準確地呈比例的結果,本研究利用450ml的波長進行了實驗。表1根據檢測波長阿雷素各濃度的吸光度效果實施例2為了檢測阿雷素吸附至明膠微粒的條件,把顆粒水化(hydration)到多種溶液里。在此所使用的溶液為包括磷酸緩沖生理鹽水(PBC)的12種緩沖液和無菌及生理鹽水(normalsaline)。表2明膠微粒水化(hydration)溶液No.溶液No.溶液1200mMTris-HClpH9.88100mMcitratepH32200mMTris-HClpH89100mMcitratepH43200mMTris-HClpH7.510100mMcitratepH54200mMTris-HClpH6.811100mMcitratepH65200mMTris-KClpH2D.W無菌水6200mMglycinepH3Saline0.85%NaCl7200mMglycinepH9.6PBS磷酸鹽緩沖鹽水pH7.5明膠微粒的水化過程概要如下。把10mL本發明的明膠顆粒分別放入表2所示的14種溶液中,并在4℃下攪拌一個晚上。把1mL的此懸浮液移到e-tube后,進行10分鐘vortexing。將其在13000rpm下進行2分鐘的離心分離之后,清除上層清液,并添加稀釋為200ug/mL的阿雷素溶液1mL,并進行10分鐘vortexing。之后,再將其在13000rpm下進行2分鐘的離心分離之后,在450nm下,檢測了上層清液的吸光度。檢測結果如下表3及圖9~10,吸光度檢測值和阿雷素的濃度值越低,其藥劑在明膠微粒當中的吸附越好。效果實施例3表3根據水化溶液種類的明膠微粒的阿雷素吸附能力(上層清液液位吸光度檢測值及阿雷素濃度)溶液吸光度阿雷素濃度(ug/mL)0.2mg/mL2.762195.0493D.W2.736193.2183Saline2.686189.6972PBS0.84159.7676110.0947.16197220.51937.0915530.86661.5281741.23887.7253552.85201.246562.842200.683170.1037.79577582.768195.471892.736193.2183102.522178.1479111.47104.0634在蒸餾水和生理鹽水進行水化的凝膠粒子上阿雷素幾乎沒有吸附,而在PBS上進行水化的凝膠粒子上則有相當數量的阿雷素吸附。但是,值得注意的是,不管緩沖液的種類,根據pH條件來左右阿雷素的吸附。Tris及glycine緩沖液,在其最強堿性值pH-9.6~9.8下,大部分的藥劑被吸附,而在pH值3~4的glycine,檸檬酸鹽緩沖液當中,幾乎沒有發生阿雷素的吸附。綜合這些結果,可以理解為pH值高的堿性狀態下易于進行吸附,而酸性狀態越強,出現吸附被抑制,滿足此條件的化學鍵可以舉離子鍵(ionicbond)。為了確認吸附到明膠微粒上的阿雷素的化學鍵是否是可逆性結合,所以把吸附有阿雷素的凝膠粒子與pH值呈中性的生理鹽水進行了反應。把因吸附藥材而呈紅色的凝膠粒子懸浮到蒸餾水上,并在13000rpm下進行2分鐘離心分離后,扔掉上層清液,并在此添加0.5mL的生理鹽水,以此把凝膠粒子再次懸浮后,進行10分鐘的vortexing。之后,再將其在13000rpm下進行2分鐘的離心分離之后,在450nm下,檢測了上層清液的吸光度。檢測結果如下表4及圖11。PBS和Tris及glycine緩沖液中被吸附到凝膠粒子當中的阿雷素被生理鹽水洗脫出來,從此確認到洗脫程度根據pH值為臨近中性的緩沖液,其被吸附的藥劑最易于分離。綜合這些觀察結果,水化緩沖液的堿性越強,對凝膠粒子的阿雷素的吸附率就越高,而與此相比,水化中使用了靠近中性的緩沖液時,被吸附的藥劑的洗脫率則高。表4洗脫至生理鹽水的阿雷素效果實施例4為了了解根據水化溶液的種類和濃度的明膠粒子所具有的吸附阿雷素的能力,把粒子水化(Hydration)到多種溶液里。在此所使用的溶液為,包括Tris緩沖液,glycine緩沖液,磷酸鈉緩沖液,D.W.,0.2mg/mL,PBS(pH7.4),不含KCl的PBSpH7.4,不含NaCl的PBSpH7.4,不含KCl和NaCl的PBSpH7.4等的磷酸緩沖生理鹽水(PBS)的11種緩沖液和無菌水以及生理鹽水(normalsaline)。表5明膠微粒水化(hydration)溶液Tris緩沖液在pH值9時,不管摩爾濃度如何,出現最強的吸附力,Tris緩沖液在pH值8時,在12.5mM和25mM下出現最強的吸附力,且從摩爾濃度越高其吸附力就越強的實驗結果而言,與Tris緩沖液呈相反的結果。磷酸鈉緩沖液pH值8在12.5mM和25mM下出現最強的吸附力,而磷酸鈉緩沖液pH值7在25mM下出現最強的吸附力。另外,BPS緩沖液為NaCl時,其吸附力最好。表6PBSph7.4和不含KCl和NaCl的PBSpH7.4的各濃度吸附力表7根據PBSpH的吸附力PBS,pH9.50.605PBS,pH4.52.500100mMTris-磷酸鹽,pH110.191效果實施例5為了了解根據水化溶液磷酸鈉緩沖液的pH值和濃度的明膠粒子的阿雷素吸附能力,把粒子水化(Hydration)至溶液里。在此所使用的溶液為1種緩沖液和無菌水以及生理鹽(NormalSaline)。表8明膠微粒水化(hydration)溶液效果實施例6對本發明實施例制得的明膠海綿和明膠微球體的微觀結構進行表征,并選用韓國專利申請KR20150107270中的明膠海綿和明膠微球體作為對照組,KR20150107270中明膠海綿及明膠微球體的原料與本申請不同,其以市售明膠海綿為起始原料,而本發明以明膠發泡膠為起始原料,具體表征結果參見附圖13-16。圖13為對照組明膠海綿的數碼照片(a)和不同放大倍率下的SEM照片(b、c)。由圖13可知,對照組的明膠海綿孔隙大小不均一,且孔隙形成得較大。圖14為對照組明膠微球體的SEM照片。圖15為本發明明膠海綿不同放大倍率下的SEM照片。由圖14可知,本發明明膠海綿的孔隙大小均一,孔結構穩定。圖16為本發明明膠微球體的顯微鏡照片(a)和SEM照片(b)。比較圖16和圖14可知,采用本發明明膠發泡膠為起始原料制得的微球體仍保持了較好的孔結構。應該理解如上述的本發明的說明只是為了例示,本發明所屬的
技術領域:
中的本領域技術人員在不變更本發明的技術構思或必要特征的情況下,能夠以其他具體方式容易進行變形。因此,應該理解以上記載的實施例是在所有方面上的例示,并不是限定性的。例如,以單一形式進行說明的各構成要素,也可以被分散地實施,同樣被分散地進行說明的構成要素也可以以結合的形式被實施。本發明的范圍不是通過
發明內容而是由后附的權利要求書來體現,應解釋為,權利要求書的含義和范圍以及由其均等的概念推導出的所有變更或變形的方式均包含在本發明的范圍內。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1