聚合物、其制備方法及其應(yīng)用與流程
本發(fā)明涉及高分子合成技術(shù)領(lǐng)域,尤其涉及一種聚合物、其制備方法及其應(yīng)用。
背景技術(shù):
大量實驗和臨床研究表明人類實體腫瘤生長于一個獨特的微環(huán)境中,在這里腫瘤細(xì)胞代謝異常旺盛并且不斷增值,然而腫瘤組織異常的血供系統(tǒng)導(dǎo)致腫瘤細(xì)胞的氧氣及養(yǎng)分供應(yīng)不足。這些乏氧細(xì)胞對放射治療有一定的抗性,不僅給腫瘤治療增加了難度而且還是腫瘤復(fù)發(fā)的根源。目前,超過50%的腫瘤病人都采用了放射治療,若用正常組織可耐受的劑量照射,腫瘤治愈率一般只能達(dá)到40%左右,但是許多腫瘤即使接受高劑量照射后仍效果欠佳。據(jù)計算,對于殺死同樣大小的腫瘤,乏氧腫瘤所需的照射劑量大約相當(dāng)于有氧腫瘤的3倍,由此可見腫瘤中的乏氧細(xì)胞對射線有較強(qiáng)的耐受性。同時,高劑量的照射也十分容易造成對正常組織細(xì)胞的損傷。因此,如何提高腫瘤中的乏氧細(xì)胞對射線的敏感性是提高放射治療效果的關(guān)鍵。
當(dāng)前,腫瘤的發(fā)病率呈逐年上升趨勢。高效的放射增敏劑的研制不僅可以有效的提高腫瘤的治愈率而且還能產(chǎn)生較大的經(jīng)濟(jì)效益和社會效益。具有腫瘤放療增敏活性的小分子放療增敏劑有很多種,但是目前獲得臨床應(yīng)用的卻很少。目前在臨床中獲得應(yīng)用或者在臨床實驗階段的,主要是硝基咪唑類化合物,它們可以將放射治療時射線對腫瘤乏氧細(xì)胞造成的DNA損傷固定并抑制其DNA的修復(fù)。其中,2-硝基咪唑類化合物雖然增敏活性較高,但是也具有較高的神經(jīng)毒性,因而未能獲得廣泛應(yīng)用。5-硝基咪唑類化合物也被證明具有較好放射增敏的性能,并且目前唯一獲得臨床批準(zhǔn)的,作為腫瘤放療增敏劑的甘氨雙唑鈉已經(jīng)上市銷售,它可以提高實體腫瘤組織內(nèi)乏氧細(xì)胞對射線的敏感性且對正常組織無明顯影響。但是甘氨雙唑鈉在水溶液中的穩(wěn)定性極差,而且在腫瘤組織中的富集效果不理想。這些缺陷將導(dǎo)致甘氨雙唑鈉進(jìn)入機(jī)體后,在短時間內(nèi)就被代謝完全。這些缺點限制了其大規(guī)模的應(yīng)用。
近年來,關(guān)于納米藥物的制備和應(yīng)用越來越引起人們的關(guān)注。由于腫瘤組織具有高通透性和滯留效應(yīng)(EPR效應(yīng)),使得聚合物納米粒子可以在腫瘤組織中高效富集。同時,具有兩親性的聚合物納米粒子能夠在機(jī)體內(nèi)的循環(huán)系統(tǒng)中長期穩(wěn)定的存在。因此,基于以上優(yōu)點,構(gòu)筑具有放射增敏性能的聚合物,并通過自組裝的方法制備成納米粒子,能夠通過血液循環(huán)到達(dá)腫瘤組織并在腫瘤組織中富集從而實現(xiàn)更加高效的放療增敏效果。
技術(shù)實現(xiàn)要素:
本發(fā)明的目的提供一種聚合物、其制備方法及其應(yīng)用,本發(fā)明提供的聚合物作為腫瘤放療增敏劑使用時,具有高效的腫瘤放療增敏效果。
本發(fā)明提供了一種式(I)所示的聚合物:
其中,m和n分別為聚合度。
在一個具體實施例中,40≤m≤250,20≤n≤200。
在一個具體實施例中,100≤m≤120,40≤n≤150。
本發(fā)明提供的聚合物含有聚乙二醇單甲醚、谷氨酸、甲硝唑等基團(tuán),為兩親性嵌段共聚物,能夠在水溶液中自組裝形成穩(wěn)定的膠束納米粒子,從而可以作為腫瘤放療增敏劑應(yīng)用。
本發(fā)明還提供了一種腫瘤放療增敏劑,由上述技術(shù)方案所述的聚合物在水溶液中自組裝形成。
在一個實施例中,所述聚合物在水溶液中的濃度為1mg/mL~10mg/mL。
在一個實施例中,所述聚合物為10nm~200nm的膠束納米粒子。
本發(fā)明還提供了上述技術(shù)方案所述的聚合物的制備方法,包括:
式(II)所示的聚乙二醇-b-聚谷氨酸丙炔酯嵌段聚合物與式(III)所示的疊氮甲硝唑進(jìn)行點擊化學(xué)反應(yīng)得到式(I)所示的聚合物;
其中,m和n分別為聚合度。
本發(fā)明中嵌段聚合物的合成方法路線如下所示,以谷氨酸和甲硝唑為主要原料,經(jīng)過一系列簡單的有機(jī)反應(yīng)以及開環(huán)聚合反應(yīng),最后通過高效的“點擊反應(yīng)”制得嵌段聚合物。
具體過程如下:
(1)首先將谷氨酸和炔丙醇反應(yīng),再通過關(guān)成環(huán)反應(yīng)得到含炔基的內(nèi)酸酐單體,進(jìn)一步以端基為氨基的聚乙二醇作為引發(fā)劑,引發(fā)開環(huán)反應(yīng),得到含炔基官能團(tuán)的兩嵌段聚合物聚乙二醇-b-聚谷氨酸丙炔酯。該合成參考文獻(xiàn)Angew.Chem.Int.Ed.2009,48,9334-9338。其中聚乙二醇和聚谷氨酸丙炔酯嵌段的長度可以通過所采用的聚乙二醇引發(fā)劑長度以及開環(huán)聚合過程中的投料比和反應(yīng)時間來控制。在一個實施例中,聚乙二醇的分子量為2000~10000,聚谷氨酸丙炔酯嵌段的聚合度為40~150。
(2)設(shè)計疊氮化的甲硝唑小分子。首先通過甲硝唑和對甲苯磺酰氯反應(yīng)活化甲硝唑中的羥基,進(jìn)一步通過與疊氮化鈉的取代反應(yīng),可以得到疊氮化的甲硝唑。
(3)將如上得到的嵌段聚合物聚乙二醇-b-聚谷氨酸丙炔酯和疊氮化的甲硝唑進(jìn)行點擊化學(xué)反應(yīng),制備得到最終的嵌段聚合物聚乙二醇-b-聚(谷氨酸-g-甲硝唑)。由于點擊化學(xué)反應(yīng)的高效性,我們只要在反應(yīng)時將甲硝唑過量3倍,就可以將炔基完全接上甲硝唑。
通過嵌段聚合物聚乙二醇-b-聚(谷氨酸-g-甲硝唑)在水溶液中進(jìn)行自組裝,可以得到以聚乙二醇為親水殼層,以聚(谷氨酸-g-甲硝唑)為疏水核心的兩親性嵌段聚合物膠束納米粒子。具體的方法是先將聚乙二醇-b-聚(谷氨酸-g-甲硝唑)溶解在乙腈溶液中,然后迅速加入水中形成穩(wěn)定的納米粒子,最后將其中的乙腈溶劑通過減壓蒸餾的方法除去。
本發(fā)明提供的兩親性嵌段共聚物在水溶液中進(jìn)行自組裝,得到以聚乙二醇為親水殼層,以聚(谷氨酸-g-甲硝唑)為疏水核心的兩親性嵌段聚合物膠束納米粒子,其可以作為腫瘤放療增敏劑使用。實驗結(jié)果表明,本發(fā)明提供的兩親性嵌段共聚物作為腫瘤放療增敏劑可以在體內(nèi)血液的環(huán)境中長時間循環(huán)并在腫瘤組織中富集,而且具有十分高效的腫瘤放療增敏效果,顯著優(yōu)于市售的甘氨雙唑鈉注射液。
附圖說明
圖1為本發(fā)明實施例制備的谷氨酸丙炔酯-N-羧基內(nèi)酸酐的1H NMR表征圖;
圖2為本發(fā)明實施例制備的聚乙二醇-b-聚谷氨酸丙炔酯嵌段聚合物的1HNMR表征圖;
圖3為本發(fā)明實施例制備的疊氮甲硝唑的1HNMR表征圖;
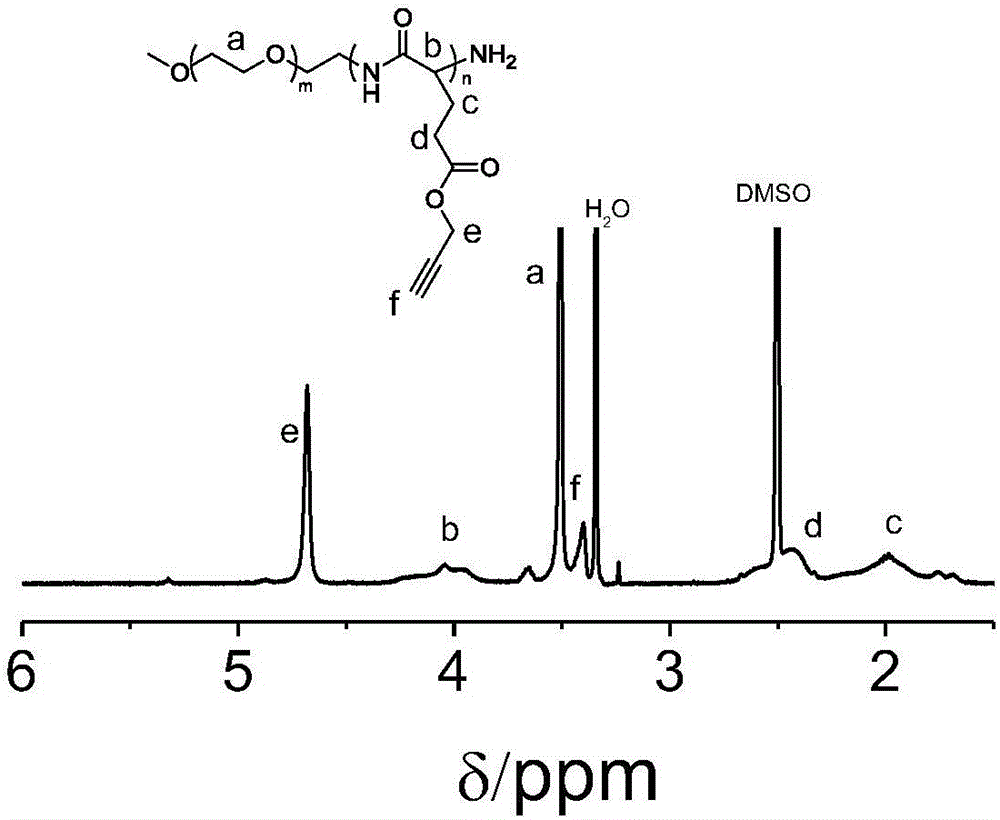
圖4為本發(fā)明實施例制備的兩親性嵌段聚合物聚乙二醇-b-(聚谷氨酸-g-甲硝唑)的1H NMR表征圖;
圖5為本發(fā)明實施例制備的兩親性嵌段聚合物聚乙二醇-b-(聚谷氨酸-g-甲硝唑)的體積排除色譜表征圖;
圖6為以芘作為熒光探針,聚合物濃度對激發(fā)光光譜中波長在339nm和332nm處的強(qiáng)度的比值圖;
圖7為濃度為1mg/mL的聚合物納米粒子溶液的動態(tài)光散射圖;
圖8為增敏劑注射后得到H22腫瘤的生長曲線;
圖9為治療后切下的腫瘤的照片。
具體實施方式
為了進(jìn)一步說明本發(fā)明,下面結(jié)合實施例對本發(fā)明提供的聚合物、其制備方法及其應(yīng)用進(jìn)行詳細(xì)地描述,但不能將它們理解為對本發(fā)明保護(hù)范圍的限定。
實施例1
1.1谷氨酸丙炔酯的制備
(1)在250mL圓底燒瓶中分別加入事先量好的谷氨酸(4g,27mmol)和丙炔醇(120mL),并放入磁子,在磁力攪拌器上攪拌0.5h,期間控制油浴溫度為40℃。
(2)用滴液漏斗滴加14mL三甲基氯硅烷,30min滴完,繼續(xù)攪拌2天。觀察顏色變化,由白色逐漸變?yōu)榘迭S色最終為紅褐色。
(3)在1L燒杯中倒入900mL無水乙醚,放在磁力攪拌器上攪拌。將反應(yīng)混合液逐滴滴加到無水乙醚中,觀察現(xiàn)象,發(fā)現(xiàn)有白色固體析出。抽慮后放入真空干燥箱中干燥過夜即可得到谷氨酸丙炔酯,為白色固體,產(chǎn)率為82%。
1.2谷氨酸丙炔酯-N-羧基內(nèi)酸酐的制備
(1)在25mL圓底燒瓶中加入上述制備的谷氨酸丙炔酯(925mg,5mmol)和二(三氯甲基)碳酸酯(BTC)(600mg,2mmol),并放入磁子。裝好冷凝管(用于回流)。裝置抽真空。氮氣保護(hù)的同時加入15mL無水四氫呋喃,油浴加熱攪拌2h,溫度控制在70℃,通冷卻水。
(2)停止加熱,溫度降至室溫。在100mL燒杯中加入80mL冷的正己烷,將反應(yīng)液滴入正己烷中,并置于-20℃環(huán)境中過夜。
(3)發(fā)現(xiàn)燒杯底部出現(xiàn)棕褐色的油狀物。將上層正己烷移走,加入50mL干燥的乙酸乙酯溶解油狀物。依次用100mL冰水、100mL冰的飽和碳酸氫鈉溶液和100mL冰的NaCl溶液洗滌,用無水硫酸鈉干燥后過濾濃縮備用。產(chǎn)率為50%。谷氨酸丙炔酯-N-羧基內(nèi)酸酐的1H NMR表征如圖1所示,其中,EA表示未完全干燥除去的溶劑乙酸乙酯。
1.3聚乙二醇-b-聚谷氨酸丙炔酯嵌段聚合物的制備
(1)在10mL圓底燒瓶中加入(200mg,0.4mmol)單端氨基聚乙二醇(分子量5000g/mol),放入磁子,加入二氯甲烷5滴溶解,另加入5mL甲苯,橡皮塞封口。放入液氮中冷凍并用真空泵除水2h。
(2)除水完畢,將(600mg,2.8mmol)谷氨酸丙炔酯-N-羧基內(nèi)酸酐用5mL無水N,N-二甲基甲酰胺溶解并用10mL注射器加入燒瓶中同時通氮氣保護(hù)。用磁力攪拌器攪拌,室溫下反應(yīng)3天。
(3)將反應(yīng)液滴加到50mL無水乙醚中,產(chǎn)生沉淀,并放入離心機(jī)中離心。移去上清液,用5mL二氯甲烷溶解離心管底部固體,重復(fù)上述沉淀操作兩次。最后放入真空干燥箱中干燥過夜。計算產(chǎn)率為90%。聚乙二醇-b-聚谷氨酸丙炔酯嵌段聚合物1H NMR表征如圖2示。
1.4疊氮甲硝唑的制備
(1)稱取甲硝唑(3.08g,18mmol)置于50mL圓底燒瓶中,另加入三乙胺3mL(溶于20mL無水二氯甲烷中),放入磁子置于磁力攪拌器上攪拌。將對甲苯磺酰氯(3.83g,20mmol)溶于10mL無水二氯甲烷,并通過滴液漏斗滴加到懸濁液中。反應(yīng)裝置放于0℃冰水浴中反應(yīng)5h。
(2)停止反應(yīng)。加入30mL冰水,攪拌5min后倒入分液漏斗中靜置分層。上層水相再用乙酸乙酯萃取(2×30mL)。將有機(jī)相合并并用飽和碳酸氫鈉溶液30mL、飽和NaCl溶液30mL洗滌。用無水硫酸鈉干燥,過濾,濃縮后放于真空干燥箱中過夜。產(chǎn)率為92%。
(3)取上步產(chǎn)物(4.88g,15mmol)和疊氮化鈉(1.3g,20mmol)置于100mL放有磁子的圓底燒瓶中,加入50mL無水N,N-二甲基甲酰胺,裝好冷凝管。放入油浴鍋中加熱攪拌4h,溫度為80℃,同時通冷卻水。
(4)反應(yīng)結(jié)束,降至室溫。倒入400mL水中,用氯仿萃取(3×100mL),有機(jī)相再用100mL水洗,無水硫酸鈉干燥,過濾,濃縮后放入真空干燥箱中過夜。產(chǎn)率80%。其1H NMR表征如圖3所示。
1.5聚乙二醇-b-(聚谷氨酸-g-甲硝唑)的制備
(1)取一10mL封管,放入小磁子,將聚乙二醇-b-聚谷氨酸丙炔酯(700mg,0.028mmol)、甲硝唑疊氮化合物(1g,5mmol)和N,N,N,N,N-五甲基二乙烯三胺(80mg,0.45mmol)溶于5mL無水N,N-二甲基甲酰胺中并加入到封管中。
(2)經(jīng)過三次冷凍-脫氣-充氮氣循環(huán)后,將稱量好的溴化亞銅(64mg,0.45mmol)加入到封管中,再循環(huán)兩次。真空條件下封上封管。放入40℃油浴鍋中攪拌,反應(yīng)10h。
(3)將封管放入液氮中凍住終止反應(yīng)。反應(yīng)混合液用四氫呋喃稀釋,過硅膠柱除去銅鹽,濃縮除去大部分溶劑。
(4)將剩余液體滴加到50mL無水乙醚中產(chǎn)生沉淀,離心,移去上清液,二氯甲烷溶解,重復(fù)上述操作兩次。最后放入真空干燥箱中干燥過夜,得到最終產(chǎn)品。產(chǎn)率96%。兩親性嵌段聚合物聚乙二醇-b-(聚谷氨酸-g-甲硝唑)的1H NMR譜圖表征如圖4所示,體積排除色譜表征如圖5所示。
1.6自組裝制備聚合物納米粒子
稱取聚乙二醇-b-(聚谷氨酸-g-甲硝唑)(1mg,0.033mmol)(以下簡稱聚合物)于2mL離心管中,加入0.5mL乙腈,超聲溶解。10mL樣品瓶中加入1mL磷酸鹽緩沖液,放入小磁子,開始攪拌。用移液槍將聚合物溶液迅速加入到樣品瓶中,然后濃縮除去乙腈,得到濃度為1mg/mL的聚合物納米粒子溶液。
按照1.6公開的方法分別獲得濃度為1、2、3、4、5mg/mL的聚合物納米粒子溶液;通過以芘作為熒光探針,分別測試上述聚合物納米粒子溶液在339nm和332nm激發(fā)光強(qiáng)度,并繪制聚合物濃度對激發(fā)光光譜中波長在339nm和332nm處的強(qiáng)度的比值圖,可以得到聚合物的臨界膠束濃度為9.1×10-3mg/mL,結(jié)果參見圖6,圖6為以芘作為熒光探針,聚合物濃度對激發(fā)光光譜中波長在339nm和332nm處的強(qiáng)度的比值圖,其中溶液中芘的濃度為1×10-7M,熒光測試的發(fā)射波長為475nm。由圖6可知,濃度為1~5mg/mL的聚合物納米粒子,在進(jìn)入血液循環(huán)且被小鼠血液稀釋約10倍后,依然能夠保持納米粒子的狀態(tài)。
參見圖7,圖7為濃度為1mg/mL的聚合物納米粒子溶液的動態(tài)光散射圖,由圖7可知,聚合物納米粒子的平均粒徑為40nm,該尺寸便于納米粒子在腫瘤組織部位富集。同時,自組裝所得的納米粒子在水溶液中形成的粒子,表面是親水性的聚乙二醇結(jié)構(gòu)。這些特征都說明此納米粒子可以在體內(nèi)血液的環(huán)境中長時間循環(huán)并在腫瘤組織中富集。
1.8作為增敏劑的應(yīng)用
通過體外培養(yǎng)H22肝癌細(xì)胞,將細(xì)胞洗滌后分散于磷酸緩沖溶液中,按照每只老鼠5×106個細(xì)胞的量,大腿內(nèi)側(cè)注射100μL細(xì)胞分散液,大約一周之后,得到H22小鼠肝癌細(xì)胞的皮下腫瘤模型,在腫瘤大小長至大約200mm3的時候,將所制備的聚合物納米粒子溶液以及甘氨雙唑鈉溶液,在相同的甲硝唑當(dāng)量下,分別通過尾靜脈注射到荷瘤小鼠體內(nèi),然后進(jìn)行電子槍射線放射治療,照射相同的劑量(4Gy)。每隔三天重復(fù)以上操作,共照射3次,照射完畢,繼續(xù)飼養(yǎng)觀察。每隔一天記錄腫瘤體積大小,結(jié)果參見圖8和圖9,圖8為增敏劑注射后得到H22腫瘤的生長曲線,其中NPs和GS中的甲硝唑當(dāng)量濃度為1.4mg/mL,每次注射100μL,每次的照射劑量為4Gy。NPs在照射前4h注射,GS按照使用說明書在照射前1h注射。分別在第1,4,7日進(jìn)行放射治療;圖9為治療后切下的腫瘤的照片。由圖8和圖9可知,納米粒子照射組(NPs)的腫瘤相比于照射之前,其體積大小明顯縮小,而甘氨雙唑鈉組(GS)腫瘤體積增大至原來的8倍。注射磷酸鹽緩沖溶液的對照組(PBS),腫瘤體積增大至原來的12倍。通過該實驗,充分證明了嵌段聚合物納米粒子作為腫瘤放療增敏劑具有十分高效的腫瘤放療增敏效果,與市場銷售的甘氨雙唑鈉注射液相比,效果顯著提高。
以上所述僅是本發(fā)明的優(yōu)選實施方式,應(yīng)當(dāng)指出,對于本技術(shù)領(lǐng)域的普通技術(shù)人員來說,在不脫離本發(fā)明原理的前提下,還可以做出若干改進(jìn)和潤飾,這些改進(jìn)和潤飾也應(yīng)視為本發(fā)明的保護(hù)范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!