新穎異噻唑-3-基和異噁唑-3-基磺酰胺化合物的制作方法
背景技術:
1、髓鞘形成是在發育過程中強烈發生的過程,盡管在整個成人cns中存在大量的少突膠質細胞前體細胞(opc),但在慢性脫髓鞘疾病中,向髓鞘形成少突膠質細胞的轉變和在裸露的軸突周圍產生恢復性髓鞘的過程受到損害。在發育過程中,髓鞘形成以非常有序的方式進行,其中opc的特征在于標記物諸如神經/膠質抗原2(ng2)和血小板衍生生長因子α(pdgfrα)的表達,分化為失去ng2和pdgfrα表達的少突膠質細胞并獲得標記物諸如髓鞘堿性蛋白(mbp)和髓鞘少突膠質細胞糖蛋白(mog)的表達。少突膠質細胞產生髓鞘是受到非常嚴格調控的過程,并且在cns中,這可以通過與軸突的相互作用來控制,這在外周神經系統中得到了很好的理解,但在中樞神經系統中卻沒有(macklin,w.b.(2010).sci.signal.3,pe32–pe32,“the?myelin?brake:when?enough?is?enough”)。髓鞘形成也可以由少突膠質細胞自身內的內部制動通過轉錄因子eb(tfeb)-puma軸或通過gpr17拮抗作用來控制(chen,y.等人.(2009).nat?neurosci?12,1398–1406,“the?oligodendrocyte-specific?gprotein-coupled?receptor?gpr17?is?a?cell-intrinsic?timer?of?myelination”)(sun,l.o.等人.(2018).cell?175,1811-1826.e21,“spatiotemporal?control?of?cnsmyelination?by?oligodendrocyte?programmed?cell?death?through?the?tfeb-pumaaxis”)。髓磷脂不僅用于保護軸突和促進神經元傳遞,而且少突膠質細胞還被證明在軸突的代謝以及維持軸突周圍的電解質平衡方面發揮重要作用(schirmer,l.等人.(2014).annneurol?75,810–828,“differential?loss?of?kir4.1?immunoreactivity?in?multiplesclerosis?lesions”)(simons,m.和nave,k.-a.(2015).cold?spring?harb?perspectbiol.22,“oligodendrocytes:myelination?and?axonal?support”)。
2、gpr17是a類孤兒g蛋白偶聯受體(gpcr)。gpcr是7結構域跨膜蛋白,該結構域跨膜蛋白經由它們與由gα、gβ、gγ亞單位組成的小異源三聚體g蛋白復合物的細胞內關聯,將細胞外配體與細胞內信號偶聯。正是gpcr與gα亞單位的偶聯協商產生了下游細胞內信號通路。已知gpr17直接與gαi/o偶聯,這導致抑制腺苷酸環化酶活性,從而導致環amp生成(camp)減少。gpr17還被證明與靶向磷脂酶c的gq/11偶聯。磷脂酶c的激活導致磷脂酰肌醇4,5-二磷酸裂解,該磷脂酰肌醇4,5-二磷酸產生三磷酸肌醇(ip3)和甘油二酯(dag)。ip3隨后與內質網上的ip3受體結合并導致細胞內鈣水平增加(hanlon,c.d.和andrew,d.j.(2015)。j?cellsci.128,3533-3542,“outside-in?signaling-a?brief?review?of?gpcr?signaling?witha?focus?on?the?drosophila?gpcr?family”)(inoue,a.等人.(2019),cell?177,1933-1947.e25,“illuminating?g-protein-coupling?selectivity?of?gpcrs”)。
3、gpr17在髓鞘形成中的作用首先是在olig1基因敲除小鼠的視神經篩選中確定的,以鑒定調節髓鞘形成的基因。發現gpr17表達僅在cns的髓鞘形成細胞中表達,而在施萬細胞(外圍神經系統的髓鞘形成細胞)中不存在。發現gpr17的表達僅在少突膠質細胞譜系細胞中表達,并在髓鞘形成少突膠質細胞中下調(chen,y.等人.(2009))。具體而言,發現gpr17表達在opc的早期以低水平存在,并且在成熟的髓鞘形成少突膠質細胞中表達下調之前,在髓鞘形成前少突膠質細胞中的表達增加(boda,e.等人.(2011),glia?59,1958–1973,“the?gpr17?receptor?in?ng2?expressing?cells:focus?on?in?vivocell?maturationand?participation?in?acute?trauma?and?chronic?damage”)(dziedzic,a.等人.(2020).int.j.mol.sci.21,1852,“the?gpr17receptor—a?promising?goal?for?therapyand?a?potential?marker?of?the?neurodegenerative?process?in?multiplesclerosis”)(fumagalli,m.等人.(2011),j?biol?chem?286,10593–10604,“phenotypicchanges,signaling?pathway,and?functional?correlates?of?gpr17-expressingneural?precursor?cells?during?oligodendrocyte?differentiation”)。gpr17敲除動物在整個cns中表現出早熟髓鞘形成,相反,轉基因小鼠在少突膠質細胞中過表達gpr17與cnp-cre(2',3'-環狀核苷酸3'-磷酸二酯酶)啟動子表現出髓鞘生成缺陷,這與髓鞘形成過程中細胞內在制動的預期一致(chen,y.等人.(2009))。此外,gpr17的丟失增強了溶血磷脂酰膽堿誘導的脫髓鞘的脫髓鞘作用后的髓鞘再生(lu,c.,dong等人.(2018),sci.rep.8,4502,“g-protein-coupled?receptor?gpr17?regulates?oligodendrocytedifferentiation?in?response?to?lysolecithin-induced?demyelination”)。因此,促進少突膠質細胞譜系細胞分化為成熟的髓鞘形成少突膠質細胞的gpr17的拮抗作用將導致脫髓鞘后髓鞘形成增加。
4、多發性硬化癥(ms)是一種慢性神經退行性疾病,其特征在于中樞神經系統(cns)中髓鞘(軸突周圍的保護性脂肪脂質層)丟失。預防髓鞘損失或裸露軸突的髓鞘再生被認為可以預防軸突變性,從而預防疾病的進展(franklin,r.j.(2002),nat?rev?neurosci?3,705–714,“why?does?remyelination?fail?in?multiple?sclerosis?”)。由于髓鞘修復對中樞神經系統的恢復作用,此類治療將有益于所有類型的ms,即復發緩解型、繼發進展型、原發進展型和進展型復發型ms。由于保留軸突的神經保護作用,修復丟失的髓鞘將減輕與ms相關聯的神經系統癥狀。
5、由于髓鞘形成在神經系統功能中起著重要作用,促進opc向少突膠質細胞分化有可能影響多種疾病,其中由于疾病本身或炎癥,已觀察到由于髓鞘形成少突膠質細胞丟失或opc向少突膠質細胞分化受阻而導致的白質缺陷/不規則。這是除gpr17表達本身發生改變的疾病之外的疾病。
6、因此,gpr17拮抗作用可用于產生陽性疾病結果的疾病包括但不限于:
7、對髓鞘的直接損傷:
8、-導致中樞髓鞘破壞的代謝病癥,諸如腦橋中央髓鞘溶解、腦橋外髓鞘溶解,由于例如但不限于酒精中毒、肝病、移植后免疫抑制的情況下低鈉血癥的過快糾正
9、-一氧化碳中毒,據報道大腦深層白質層出現少突膠質細胞功能障礙
10、和再生障礙
11、-營養缺乏,導致髓鞘損失或在發育過程中無法正確生成髓鞘
12、-病毒誘導的脫髓鞘
13、原發性脫髓鞘疾病
14、-多發性硬化癥(復發緩解型、繼發進展型、原發進展型和進展型復發型ms)
15、-急性和多相播散性腦脊髓炎
16、-視神經脊髓炎譜系疾病,包括視神經炎
17、-橫貫性脊髓炎
18、-腦白質營養不良,諸如腎上腺腦白質營養不良、腎上腺髓鞘神經病和其他導致髓鞘損失的遺傳性腦白質營養不良
19、具有相關聯的髓鞘損失的cns疾病:
20、-阿爾茨海默病
21、-精神分裂癥
22、-帕金森病
23、-亨廷頓病
24、-肌萎縮性側索硬化
25、-卒中引起的缺血
26、其他疾病:
27、-cns中的炎癥例如腦炎、原發性血管炎、腦膜炎后cns中的炎癥式i化合物結合gpr17并調節其活性。
28、因此,式i化合物特別可用于治療與gpr17拮抗作用相關的疾病。
29、式i化合物特別可用于治療或預防多發性硬化癥(ms)、與以下相關的病癥:髓鞘直接損傷(諸如一氧化碳中毒或病毒誘導的脫髓鞘)、原發性脫髓鞘疾病(諸如急性和多相播散性腦脊髓炎)、以及其他與髓鞘損失相關聯的cns疾病(諸如阿爾茨海默病、精神分裂癥、帕金森病和亨廷頓病)。
技術實現思路
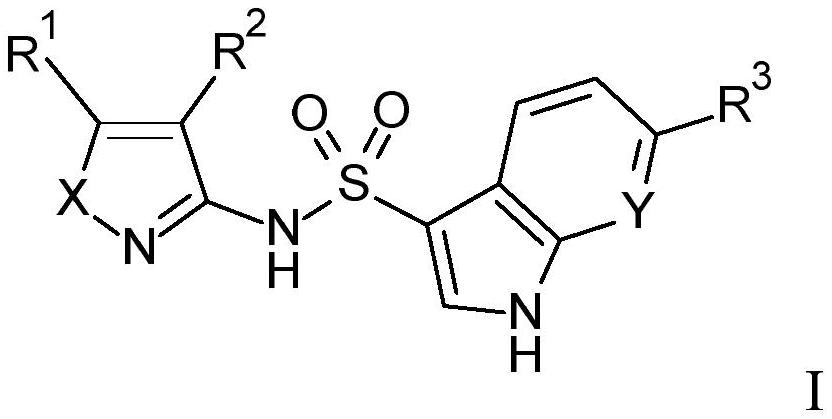
1、本發明提供式i的新穎化合物,
2、
3、其中,
4、r1為烷基、烷氧基、氰基、氰基烷基、環丙基或鹵代;
5、r2為h、烷基、烷氧基、氰基、環丙基或鹵代;
6、r3為鹵代、烷基或鹵代烷基;
7、x為–o–或–s-;
8、y為ch或n;
9、及其藥用鹽。
10、術語“烷基”表示具有1至6個碳原子的單價直鏈或支鏈飽和烴基。在一些實施例中,如果沒有另行說明,則烷基包含1個至6個碳原子(c1-6-烷基)或1個至4個碳原子(c1-4-烷基)。c1-6-烷基的實例包括甲基、乙基、丙基、異丙基、正丁基、異丁基、仲丁基、叔丁基和戊基。特定的烷基基團包括甲基、乙基和丙基。當指出具有特定碳數的烷基殘基時,可涵蓋具有該碳數的所有幾何異構體。因此,例如“丁基”可包括正丁基、仲丁基、異丁基和叔丁基,并且“丙基”可包括正丙基和異丙基。
11、術語“烷氧基”表示式-o-r'的基團,其中r'為c1-6-烷基基團。c1-6-烷氧基基團的實例包括甲氧基、乙氧基、正丙氧基、異丙氧基、正丁氧基、異丁氧基和叔丁氧基。特定實例為甲氧基和乙氧基。
12、術語“氰基”表示-c≡n基團。
13、“氰基烷基”意指式-r'-r"的部分,其中r'是如本文所定義的烷基并且r"是氰基或腈基。特定的實例為氰基乙基。
14、術語“鹵素”、“鹵化物”和“鹵代”在本文中可互換使用并且表示氟、氯、溴或碘。特定的鹵素為氯和溴。
15、術語“藥用鹽”是指保留游離堿或游離酸的生物效果和性質的那些鹽,這些鹽在生物學或其他方面不是不合需要的。這些鹽用無機酸諸如鹽酸、氫溴酸、硫酸、硝酸、磷酸(特別是鹽酸)和有機酸諸如甲酸、乙酸、丙酸、乙醇酸、丙酮酸、草酸、馬來酸、丙二酸、琥珀酸、富馬酸、酒石酸、檸檬酸、苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、對甲苯磺酸、水楊酸、n-乙酰基半胱氨酸形成。另外,這些鹽可通過將無機堿或有機堿加入游離酸中來制備。衍生自無機堿的鹽包括但不限于鈉鹽、鉀鹽、鋰鹽、銨鹽、鈣鹽、鎂鹽。衍生自有機堿的鹽包括但不限于以下物質的鹽:伯胺、仲胺和叔胺,包括天然存在的取代胺的取代胺,環胺和堿性離子交換樹脂,如異丙胺、三甲胺、二乙胺、三乙胺、三丙胺、乙醇胺、賴氨酸、精氨酸、n-乙基哌啶、哌啶、多胺樹脂。式i化合物也可以兩性離子的形式存在。特別優選的式i化合物的藥用鹽是與甲酸形成的鹽和與鹽酸形成的鹽,產生鹽酸鹽、二鹽酸鹽或三鹽酸鹽。
16、縮寫um意指微摩爾,并且與符號μm等同。
17、縮寫ul意指微升,并且與符號μl等同。
18、縮寫ug意指微克,并且與符號μg等同。
19、式i化合物可以含有若干不對稱中心,并且可以以光學純對映體、對映體的混合物(例如外消旋體)、光學純非對映體、非對映體的混合物、非對映外消旋體或非對映外消旋體的混合物存在。
20、根據cahn-ingold-prelog規范,不對稱碳原子可以是"r"或"s"構型。
21、本發明的另一實施例提供如本文所述的根據式i的化合物及其藥用鹽或酯,特別是如本文所述的根據式i的化合物及其藥用鹽,更特別地是如本文所述的根據式i的化合物。
22、本發明的實施例提供了如本文所述的根據式i化合物,其中r1為烷基、烷氧基、氰基烷基或環丙基。
23、本發明的實施例提供了如本文所述的根據式i化合物,其中r2為h、烷基、烷氧基、氰基、環丙基或鹵代。
24、本發明的實施例提供了如本文所述的根據式i化合物,其中r2為烷氧基或鹵代。
25、本發明的實施例提供了如本文所述的根據式i化合物,其中r3為鹵代或烷基。
26、本發明的實施例提供了如本文所述的根據式i化合物,其中r3為鹵代。
27、本發明的實施例提供了如本文所述的根據式i化合物,其中x為–s-。
28、本發明的實施例提供了如本文所述的根據式i化合物,其中
29、r1為烷基、烷氧基、氰基烷基或環丙基;
30、r2為h、烷基、烷氧基、氰基、環丙基或鹵代;
31、r3為鹵代;
32、x為–o–或–s-;
33、y為ch或n;
34、及其藥用鹽。
35、本發明的實施例提供了如本文所述的根據式i化合物,其中
36、r1為烷基、烷氧基、氰基烷基或環丙基;
37、r2為烷氧基或鹵代;
38、r3為鹵代;
39、x為–s-;
40、y為ch或n;
41、及其藥用鹽。
42、如本文所述的式i化合物的特別的實例選自
43、6-氯-n-(4-氯-5-丙基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
44、6-氯-n-(4-氯-5-環丙基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
45、6-氯-n-(4-氯-5-環丙基-異噻唑-3-基)-1h-吡咯并[2,3-b]吡啶-3-磺酰胺;
46、6-溴-n-(4-氯-5-環丙基-異噻唑-3-基)-1h-吡咯并[2,3-b]吡啶-3-磺酰胺;
47、6-氯-n-(4-氯-5-甲基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
48、6-氯-n-(4,5-二甲基異噻唑-3-基)-1h-吲哚-3-磺酰胺;
49、6-氯-n-(5-乙基-4-甲基-異噁唑-3-基)-1h-吲哚-3-磺酰胺;
50、6-氯-n-(4-氯-5-乙基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
51、6-氯-n-(4-乙基-5-甲基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
52、6-氯-n-(4-氯-5-甲氧基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
53、6-氯-n-(4-氯-5-乙氧基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
54、6-氯-n-[4-氯-5-(2-氰基乙基)異噻唑-3-基]-1h-吲哚-3-磺酰胺;
55、6-氯-n-(5-甲基異噻唑-3-基)-1h-吲哚-3-磺酰胺;
56、6-氯-n-(4-環丙基-5-甲基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
57、6-氯-n-(4-氰基-5-甲基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
58、6-氯-n-(4-甲氧基-5-甲基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
59、6-氯-n-(4-甲氧基-5-丙基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
60、6-氯-n-(4-甲基-5-丙基-異噁唑-3-基)-1h-吲哚-3-磺酰胺;
61、6-氯-n-(4,5-二甲基異噁唑-3-基)-1h-吲哚-3-磺酰胺;
62、6-氯-n-(4-氯-5-甲基-異噁唑-3-基)-1h-吲哚-3-磺酰胺;
63、n-(4-溴-5-甲基-異噁唑-3-基)-6-氯-1h-吲哚-3-磺酰胺;
64、及其藥用鹽。
65、如本文所述的式i化合物的優選的實例選自
66、6-氯-n-(4-氯-5-丙基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
67、6-氯-n-(4-氯-5-環丙基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
68、6-溴-n-(4-氯-5-環丙基-異噻唑-3-基)-1h-吡咯并[2,3-b]吡啶-3-磺酰胺;
69、6-氯-n-(4-氯-5-甲基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
70、6-氯-n-(4-氯-5-乙基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
71、6-氯-n-(4-氯-5-乙氧基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
72、6-氯-n-[4-氯-5-(2-氰基乙基)異噻唑-3-基]-1h-吲哚-3-磺酰胺;
73、6-氯-n-(4-甲氧基-5-丙基-異噻唑-3-基)-1h-吲哚-3-磺酰胺;
74、及其藥用鹽。
75、制造如本文所述的式i化合物的方法是本發明的目的。
76、本發明的式i化合物及其藥用鹽可以通過本領域中已知的方法,例如通過下述方法制備,該方法包括在選自n-乙基二異丙胺或吡啶的堿的存在下在添加或不添加催化劑4-二甲基氨基吡啶的情況下,或者在無機堿如磷酸鉀的存在下,使式iii化合物與式ii化合物反應以提供式i化合物,
77、
78、其中r1、r2、r3、x和y如上所述。
79、一般合成方案式i化合物可根據上述方法變體和以下方案1制備。起始材料是可商業獲得的或可以根據已知方法制備。
80、方案1
81、
82、通式i化合物可以通過在堿如n-乙基二異丙胺或吡啶的存在下在添加或不添加催化量的4-二甲基氨基吡啶的情況下,或者在無機堿如磷酸鉀的存在下,使磺酰氯ii與胺iii反應來制備。起始材料是可商業獲得的或可根據已知方法制備(參見方案1)。
83、方案2
84、
85、式iii的胺可以通過酯iv與堿(諸如氫氧化鈉或氫氧化鋰)皂化以得到酸v來制備。酸v與疊氮磷酸二苯酯在叔丁醇存在下反應生成中間體vi,其可以通過用酸(諸如鹽酸或三氟乙酸)處理脫保護來得到胺iii(參見方案2)。
86、本發明的另一實施例提供一種含有本發明化合物和治療惰性載體、稀釋劑或賦形劑的藥物組合物或藥物,以及使用本發明化合物來制備此類組合物和藥物的方法。在一個實例中,式i化合物可通過在環境溫度在適當的ph和期望的純度下與生理學上可接受的載體(即在所用劑量和濃度下對接受者無毒的載體)混合而配制為蓋倫(galenical)施用形式。配制物的ph主要取決于化合物的具體用途和濃度,但優選地在約3至約8的范圍內。在一個實例中,將式i化合物在ph?5的乙酸鹽緩沖液中配制。在另一實施例中,式i化合物為無菌的。化合物可以例如作為固體或無定形組合物、作為凍干配制物或作為水溶液儲存。
87、以與良好醫學實踐一致的方式配制、計量和施用組合物。在這種情況下需要考慮的因素包括所治療的特定疾患、所治療的特定哺乳動物、個體患者的臨床病癥、疾患的原因、藥劑的遞送部位、施用方法、施用的時間安排,以及執業醫師已知的其他因素。
88、本發明的化合物可通過任何適合的方式施用,包括口服、局部(包括頰和舌下)、直腸、陰道、經皮、腸胃外、皮下、腹膜內、肺內、皮內、鞘內和硬膜外和鼻內,以及(如果需要用于局部治療)病灶內施用。腸胃外輸注包括肌內、靜脈內、動脈內、腹膜內或皮下施用。
89、本發明化合物可以任何方便的施用形式施用,例如,片劑、粉劑、膠囊劑、溶液劑、分散劑、混懸劑、糖漿劑、噴霧劑、栓劑、凝膠劑、乳劑、貼劑等。此類組合物可以包含藥物制劑中常規的組分,例如,稀釋劑、載體、ph調節劑、甜味劑、填充劑和其他活性劑。
90、通過混合本發明的化合物和載體或賦形劑來制備通常的配制物。適合的載體和賦形劑為本領域技術人員熟知的,并且在例如ansel,howard?c.等人,ansel’spharmaceutical?dosage?forms?and?drug?delivery?systems.philadelphia:lippincott,williams和wilkins,2004;gennaro,alfonso?r.等人remington:the?scienceand?practice?of?pharmacy.philadelphia:lippincott,williams和wilkins,2000;以及rowe,raymond?c.handbook?of?pharmaceutical?excipients.chicago,pharmaceuticalpress,2005中有詳細描述。配制物還可以包含一種或多種緩沖劑、穩定劑、表面活性劑、潤濕劑、潤滑劑、乳化劑、懸浮劑、防腐劑、抗氧化劑、不透明劑、助流劑、加工助劑、著色劑、甜味劑、加香劑、調味劑、稀釋劑和其他已知的添加劑,以提供美觀的藥物(例如,本發明的化合物或其藥物組合物)展示或有助于藥物產品(例如,藥物)的制備。
91、式i化合物及其藥用鹽可與藥學上惰性的無機或有機助劑一起加工,以用于生產片劑、包衣片劑、糖衣丸、硬明膠膠囊、注射液或局部制劑。例如,可使用乳糖、玉米淀粉或其衍生物、滑石粉、硬脂酸或其鹽等作為片劑、糖衣丸和硬明膠膠囊的此類助劑。
92、用于軟明膠膠囊的合適的助劑為例如植物油、蠟、脂肪、半固體物質和液體多元醇等。
93、用于制備溶液和糖漿的合適的助劑為例如水、多元醇、蔗糖、轉化糖、葡萄糖等。
94、用于注射液的合適的助劑為例如水、醇、多元醇、甘油、植物油等。
95、用于栓劑的合適的助劑為例如天然或硬化油、蠟、脂肪、半固體或液體多元醇等。
96、用于局部眼用制劑的合適的助劑為例如環糊精、甘露醇或本領域已知的許多其他載體和賦形劑。
97、此外,藥物制劑可以含有防腐劑、增溶劑、增粘物質、穩定劑、潤濕劑、乳化劑、甜味劑、著色劑、香料、用于改變滲透壓的鹽、緩沖劑掩模劑或抗氧化劑。它們還可以含有其他有治療價值的物質。
98、劑量可以在寬范圍內變化,當然將適合每種特定情況下的各種要求。一般而言,在口服施用的情況下,每kg體重約0.1mg至20mg、優選地每kg體重約0.5mg至4mg(例如每人約300mg)的日劑量應當是合適的,其優選地分為1至3個單獨的劑量(可由例如相同的量組成)。在局部施用的情況下,配制物可包含按重量計0.001%至15%的藥物,并且所需劑量可以為0.1mg至25mg,每天或每周單次施用、或每天多次施用(2至4次),或每周多次施用。但是,顯而易見的是,當顯示為適用時,可超過本文中給出的上限或下限。
99、本發明還特別涉及:
100、一種用作治療活性物質的式i化合物;
101、用于治療由gpr17調節的疾病的式i化合物;
102、同樣,本發明的一目的為一種藥物組合物,其包含如本文所述的根據式i的化合物以及治療惰性載體。
103、式i化合物用于治療或預防病癥的用途,所述病癥由髓鞘直接損傷(包括但不限于腦橋中央和腦橋外髓鞘溶解、一氧化碳中毒、營養缺乏和病毒誘導的脫髓鞘)、脫髓鞘疾病(包括但不限于多發性硬化癥、急性和多相播散性腦脊髓炎、視神經脊髓炎譜系疾病和腦白質營養不良)、與髓鞘損失相關的cns疾病(包括但不限于阿爾茨海默病、精神分裂癥、帕金森病、亨廷頓病、肌萎縮性側索硬化癥和卒中引起的缺血)以及cns中的炎癥例如腦炎、原發性血管炎、腦膜炎和肥胖癥后cns中的炎癥引起。
104、本發明的實施例是式i化合物用于治療或預防多發性硬化癥、阿爾茨海默病、帕金森病或亨廷頓病的用途。
105、本發明的特定實施例是式i化合物用于治療或預防多發性硬化癥的用途。
106、式i化合物用于制備用于治療或預防病癥的藥物用途,所述病癥由髓鞘直接損傷(包括但不限于腦橋中央和腦橋外髓鞘溶解、一氧化碳中毒、營養缺乏和病毒誘導的脫髓鞘)、脫髓鞘疾病(包括但不限于多發性硬化癥、急性和多相播散性腦脊髓炎、視神經脊髓炎譜系疾病和腦白質營養不良)、與髓鞘損失相關的cns疾病(包括但不限于阿爾茨海默病、精神分裂癥、帕金森病、亨廷頓病、肌萎縮性側索硬化癥和卒中引起的缺血)以及cns中的炎癥例如腦炎、原發性血管炎、腦膜炎和肥胖癥后cns中的炎癥引起。
107、本發明的實施例是式i化合物用于制備用于治療或預防多發性硬化癥、阿爾茨海默病、帕金森病或亨廷頓病的藥物的用途。
108、本發明的特定實施例是式i化合物用于制備用于治療或預防多發性硬化癥的藥物的用途。
109、根據式i的化合物,其用于治療或預防病癥,所述病癥由髓鞘直接損傷(包括但不限于腦橋中央和腦橋外髓鞘溶解、一氧化碳中毒、營養缺乏和病毒誘導的脫髓鞘)、脫髓鞘疾病(包括但不限于多發性硬化癥、急性和多相播散性腦脊髓炎、視神經脊髓炎譜系疾病和腦白質營養不良)、與髓鞘損失相關的cns疾病(包括但不限于阿爾茨海默病、精神分裂癥、帕金森病、亨廷頓病、肌萎縮性側索硬化癥和卒中引起的缺血)以及cns中的炎癥例如腦炎、原發性血管炎、腦膜炎和肥胖癥后cns中的炎癥引起。
110、本發明的實施例是式i化合物,其用于治療或預防多發性硬化癥、阿爾茨海默病、帕金森病或亨廷頓病。
111、本發明的特定實施例是根據式i的化合物,其用于治療或預防多發性硬化癥。
112、一種用于治療或預防病癥的方法,所述方法包括向有此需要的患者施用有效量的式i化合物,所述病癥由髓鞘直接損傷(包括但不限于腦橋中央和腦橋外髓鞘溶解、一氧化碳中毒、營養缺乏和病毒誘導的脫髓鞘)、脫髓鞘疾病(包括但不限于多發性硬化癥、急性和多相播散性腦脊髓炎、視神經脊髓炎譜系疾病和腦白質營養不良)、與髓鞘損失相關的cns疾病(包括但不限于阿爾茨海默病、精神分裂癥、帕金森病、亨廷頓病、肌萎縮性側索硬化癥和卒中引起的缺血)以及cns中的炎癥例如腦炎、原發性血管炎、腦膜炎和肥胖癥后cns中的炎癥引起。
113、本發明的實施例是用于治療或預防多發性硬化癥、阿爾茨海默病、帕金森病或亨廷頓病的方法,該方法包括向有此需要的患者施用有效量的式i化合物。
114、本發明的特定實施例是用于治療或預防多發性硬化癥的方法,該方法包括向有此需要的患者施用有效量的式i化合物。
115、當根據所述方法中的任一者制造時,本發明的一個實施例還提供如本文所述的式i化合物。
116、測定程序
117、gpr17?camp測定方案:
118、在補充有10%胎牛血清和400μg/ml遺傳霉素的dmem(達爾伯克(氏)改良伊格爾(氏)培養基):f-12(1:1)中在37℃/5%?co2下培養穩定表達含有未標記的人gpr17縮寫(roche)的cho-k1細胞。
119、使用nano-trf檢測分析試劑盒(roche?diagnostics,cat.no.05214386001)對細胞內環磷酸腺苷(camp)水平的變化進行定量。該測定允許在均質溶液中直接對camp進行定量。基于時間分辨熒光能量轉移(tr-fret)和釕化camp和內源性camp與用alexafluor-700標記的抗camp單克隆抗體的競爭性結合來檢測camp。釕絡合物充當fret供體并將能量轉移到alexafluor-700。fret信號與camp濃度成反比。
120、用accutase分離cho-gpr17s細胞并重懸于由hank平衡鹽溶液(hbss)、10mm?hepes(4-(2-羥乙基)哌嗪-1-乙磺酸溶液)和0.1%牛血清白蛋白(ph?7.4)組成的測定緩沖液中。然后將這些細胞以每孔10’000個細胞/20μl測定緩沖液的密度接種在黑色384孔板(corning)中,直到添加化合物。
121、測試拮抗劑化合物在二甲基亞砜(dmso)中連續稀釋并點樣在384孔板中。然后將化合物稀釋在補充有ec80濃度的mdl29,951(3-(2-羧基-4,6-二氯吲哚-3-基)丙酸)(gpr17激動劑)和3-異丁基-1-甲基黃嘌呤(ibmx)的hbss緩沖液中(0.5mm最終濃度)并在室溫下添加到細胞中。在測試化合物加入5分鐘后加入毛喉素(15μm最終濃度),并將細胞在室溫下孵育30分鐘。通過在室溫下添加camp檢測混合物(含有用于細胞裂解的去污劑)90分鐘來停止測定。
122、使用paradigm讀數器(molecular?devices)測量細胞camp。根據camp試劑盒的說明,使用原始數據計算基于測定的p因子的fret信號。將數據歸一化為參考拮抗劑的最大活性,并使用s形劑量反應模型(genedata?screener)將劑量反應曲線擬合為測試化合物的活性百分比。
123、表1中提供了式i化合物的hgpr17?camp測定結果
124、表1:
125、
126、現在將通過以下實例說明本發明,所述實例不具有限制性。
127、如果制備例以對映異構體的混合物的形式獲得,則純對映異構體可通過本文所述的方法或本領域的技術人員所知的方法諸如例如手性色譜或結晶來獲得。
128、實例
129、如果沒有另行說明,則所有實例和中間體在氮氣氣氛下制備。
130、中間體a
131、中間體a1:6-氯-1h-吲哚-3-磺酰氯
132、
133、中間體a1是商業的(cas1216060-79-5)
134、中間體a2:6-氯-1h-吡咯并[2,3-b]吡啶-3-磺酰氯
135、
136、中間體a2是已知的(cas2231234-21-0)并且根據wo2018/122232合成。中間體a3:6-溴-1h-吡咯并[2,3-b]吡啶-3-磺酰氯
137、
138、中間體a3是已知的(cas2231234-27-6)并且根據wo2018/122232合成。
139、中間體b
140、中間體b1:4-氯-5-丙基-異噻唑-3-胺鹽酸鹽
141、
142、步驟1:4-氯-5-丙-1-烯基-異噻唑-3-甲酸甲酯
143、
144、將4,5-二氯異噻唑-3-甲酸甲酯(cas166668-76-4,200mg,0.94mmol)、4,4,5,5-四甲基-2-(丙-1-烯-1-基)-1,3,2-二氧雜環戊硼烷(792mg,4.72mmol)、1,1'-雙(二苯基膦基)二茂鐵-二氯化鈀(ii)二氯甲烷絡合物(39mg,0.047mmol)和碳酸鉀(521mg,3.77mmol)在1,4-二噁烷(4ml)和水(0.5ml)中的反應混合物在90℃下攪拌3h。將反應混合物用水稀釋并用乙酸乙酯(3x?5ml)萃取。將合并的有機層用水、鹽水洗滌,經硫酸鈉干燥,過濾并且濃縮至干。將殘余物通過快速色譜法(硅膠,庚烷中的0-20%乙酸乙酯)純化,以得到為淺黃色固體的4-氯-5-丙-1-烯基-異噻唑-3-甲酸甲酯(114mg,53%產率)。ms?m/z:218.0[m+h]+,esi?pos。
145、步驟2:4-氯-5-丙基-異噻唑-3-甲酸甲酯
146、
147、將4-氯-5-丙-1-烯基-異噻唑-3-甲酸甲酯(114mg,0.50mmol)在甲醇(3ml)中的溶液用氬氣吹掃并在真空下抽真空(3x)。然后添加氧化鉑(iv)(7mg,0.025mmol)并重復抽真空。在室溫下,將反應混合物在氫氣氣氛下攪拌16h。
148、將反應混合物通過硅藻土過濾并濃縮至干,以得到為灰白色液體的4-氯-5-丙基-異噻唑-3-甲酸甲酯(33mg,24%)。ms?m/z:220.1[m+h]+,esi?pos。
149、步驟3:4-氯-5-丙基-異噻唑-3-甲酸
150、
151、向4-氯-5-丙基-異噻唑-3-甲酸甲酯(33mg,0.15mmol)在四氫呋喃(0.5ml)中的溶液中添加氫氧化鈉溶液(水中的1m,179ul,0.179mmol)。將反應混合物在室溫下攪拌1h,然后用1m鹽酸酸化并用乙酸乙酯(3x?10ml)萃取。合并的有機層用水、鹽水洗滌,經硫酸鈉干燥,過濾并濃縮,以得到為淺棕色固體的4-氯-5-丙基-異噻唑-3-甲酸(37mg,98%產率)。msm/z:206.1[m+h]+,esi?pos。
152、步驟4:n-(4-氯-5-丙基-異噻唑-3-基)氨基甲酸叔丁酯
153、
154、向4-氯-5-丙基-異噻唑-3-甲酸(37.5mg,0.145mmol)在叔丁醇(0.5ml)中的溶液中添加疊氮磷酸二苯酯(44mg,34ul,0.160mmol)和三乙胺(29mg,41ul,0.29mmol)。將反應混合物在90℃下攪拌2h。冷卻至室溫后,將反應混合物用水稀釋并用乙酸乙酯(3x?10ml)萃取。將合并的有機層用水、鹽水洗滌,經硫酸鈉干燥,過濾并且濃縮至干。將粗制化合物通過快速色譜法(硅膠,庚烷中的0-25%乙酸乙酯)純化,以得到為無色油狀物的n-(4-氯-5-丙基-異噻唑-3-基)氨基甲酸叔丁酯(37mg,83%產率)。221.0[m-異丁烯+h]+,esi?pos。
155、步驟5:4-氯-5-丙基-異噻唑-3-胺鹽酸鹽
156、
157、將n-(4-氯-5-丙基-異噻唑-3-基)氨基甲酸叔丁酯(37mg,0.134mmol)在二氯甲烷(1ml)中的溶液用4m鹽酸(202mg,168ul,0.670mmol)處理,并且將混合物在23℃下攪拌1h。將反應混合物蒸發至干,以得到為白色粉末的標題化合物(20.5mg,61%)。ms?m/z:176.9[m+h]+,esi?pos。
158、中間體b2:4-甲氧基-2-丙基-嘧啶-5-胺鹽酸鹽
159、
160、步驟1:4-氯-5-環丙基-異噻唑-3-甲酸甲酯
161、
162、將4,5-二氯異噻唑-3-甲酸甲酯(cas166668-76-4,200mg,0.943mmol)、環丙基(三氟)硼酸鉀(698mg,4.72mmol)、乙酸鈀(ii)(11mg,0.047mmol)、二(金剛烷-1-基)(丁基)膦(17mg,0.047mmol)和碳酸銫(922mg,2.83mmol)在甲苯(3.5ml)和水(0.35ml)中的反應混合物在100℃下攪拌16h。將反應混合物用水稀釋并用乙酸乙酯(2x?20ml)萃取。將合并的有機層用水、鹽水洗滌,經硫酸鈉干燥,過濾并且濃縮至干。將殘余物通過快速色譜法(硅膠,庚烷中的0-25%乙酸乙酯)純化,以得到為淺黃色油狀物的4-氯-5-環丙基-異噻唑-3-甲酸甲酯(111mg,51%產率)。ms?m/z:218.0[m+h]+,esi?pos。
163、步驟2:4-氯-5-環丙基-異噻唑-3-甲酸
164、
165、向4-氯-5-環丙基-異噻唑-3-甲酸甲酯(111mg,0.484mmol)在四氫呋喃(2ml)中的溶液中添加1m氫氧化鈉水溶液(581ul,0.581mmol)。將反應混合物在室溫下攪拌1h。將反應混合物用1m鹽酸水溶液酸化并用乙酸乙酯(3x?10ml)萃取。合并的有機層用水、鹽水洗滌,經硫酸鈉干燥,過濾并濃縮,以得到為淺棕色粉末的4-氯-5-環丙基-異噻唑-3-甲酸(97mg,97%)。ms?m/z:204.0[m+h]+,esi?pos。
166、步驟3:n-(4-氯-5-環丙基-異噻唑-3-基)氨基甲酸叔丁酯
167、
168、向4-氯-5-環丙基-異噻唑-3-甲酸(97mg,0.471mmol)在叔丁醇(3ml)中的溶液中添加疊氮磷酸二苯酯(142mg,111ul,0.518mmol)和三乙胺(95mg,131ul,0.941mmol)。將反應混合物在90℃下攪拌2h,并且冷卻至室溫。將反應混合物用水稀釋并用乙酸乙酯(3x10ml)萃取。將合并的有機層用水、鹽水洗滌,經硫酸鈉干燥,過濾并且濃縮至干。將殘余物通過快速色譜法(硅膠,庚烷中的0-25%乙酸乙酯)純化,以得到為白色粉末的n-(4-氯-5-環丙基-異噻唑-3-基)氨基甲酸叔丁酯(108mg,80%)。1h?nmr(300mhz,cdcl3)δppm?0.78-0.87(m,2h)1.14-1.23(m,2h)1.54(s,9h)2.08-2.18(m,1h)7.12(br?s,1h)。步驟4:4-甲氧基-2-丙基-嘧啶-5-胺鹽酸鹽
169、
170、將n-(4-氯-5-環丙基-異噻唑-3-基)氨基甲酸叔丁酯(95mg,0.328mmol)在二氯甲烷(1ml)中的溶液用4m鹽酸水溶液(493mg,411ul,1.64mmol)處理并在室溫下攪拌1h。將反應混合物蒸發至干,以得到為白色粉末的(4-氯-5-環丙基-異噻唑-3-基)胺鹽酸鹽(76mg,99%)。ms?m/z:175.0[m+h]+,esi?pos。
171、中間體b3:4-氯-5-甲基-異噻唑-3-胺
172、
173、步驟1:4,5-二氯異噻唑-3-甲酸甲酯
174、
175、在0℃下在5min內在氬氣下向4,5-二氯異噻唑-3-甲酸(cas131947-13-2,400mg,2.02mmol)在四氫呋喃(10ml)和甲醇(1ml)中的溶液中滴加2m重氮甲基(三甲基)硅烷二乙醚(1.5ml,3.0mmol)。將淺黃色溶液在室溫下攪拌15min。將反應冷卻至0℃并用乙酸和水的溶液(1:1,0.4ml)淬滅。將黃色溶液在0℃下攪拌30min以及在室溫下攪拌約15min。添加水,并且將水相用乙酸乙酯萃取(3x)。將合并的有機層用鹽水洗滌,經硫酸鈉干燥,過濾并且在真空中濃縮。將殘余物通過快速色譜法(硅膠,庚烷中的0-20%乙酸乙酯)純化,以獲得為白色固體的4,5-二氯異噻唑-3-甲酸甲酯(395mg,92%產率)。ms?m/z:212.0[m+h]+,esi?pos。
176、步驟2:4-氯-5-甲基-異噻唑-3-甲酸
177、
178、向4,5-二氯異噻唑-3-甲酸甲酯(310mg,1.45mmol)、甲基硼酸(893mg,14.5mmol)和碳酸鉀(2.0g,14.5mmol)在1,4-二噁烷(6ml)和水(0.6ml)的混合物中的懸浮液中添加1,1'-雙(二苯基膦基)二茂鐵-二氯化鈀(ii)二氯甲烷絡合物(120mg,0.145mmol)。將橙色懸浮液在90℃下攪拌75min,然后冷卻至室溫。將橙色懸浮液用水和乙酸乙酯稀釋。將混合物用鹽酸酸化并用乙酸乙酯萃取(3x)。將有機層經硫酸鈉干燥,過濾并且在真空中濃縮,以得到為橙色固體的粗制4-氯-5-甲基-異噻唑-3-甲酸(185mg,45%純度,29%產率),其直接用于下一步驟。ms?m/z:178.1[m+h]+,esi?pos。
179、步驟3:4-氯-5-甲基-異噻唑-3-胺
180、
181、向4-氯-5-甲基-異噻唑-3-甲酸(185mg,45%純度,0.469mmol)在叔丁醇(1ml)中的溶液中添加三乙胺(95mg,131ul,0.937mmol)和疊氮磷酸二苯酯(142mg,111ul,0.516mmol)。在90℃下攪拌過夜后,添加更多的三乙胺(47mg,65ul,0.468mmol)和疊氮磷酸二苯酯(129mg,100ul,0.468mmol),并且繼續攪拌2h,然后允許反應冷卻至室溫并在真空中濃縮。將反應物用飽和碳酸氫鈉溶液稀釋并用乙酸乙酯萃取3次。合并的有機層經硫酸鈉干燥,過濾并且在真空中濃縮。將殘余物通過快速色譜法(硅膠,庚烷中的0-20%乙酸乙酯)純化,以得到為白色固體的n-(4-氯-5-甲基-異噻唑-3-基)氨基甲酸叔丁酯(20mg,17%產率)和4-氯-5-甲基-異噻唑-3-胺(16mg,23%產率)。
182、將n-(4-氯-5-甲基-異噻唑-3-基)氨基甲酸叔丁酯(20mg,0.081mmol)溶解在1,2-二氯乙烷(0.17ml)和三氟乙酸(46mg,31ul,0.404mmol)中,以獲得第二批標題化合物。在室溫下攪拌8h后將反應物用飽和碳酸氫鈉溶液淬滅并用二氯甲烷萃取(3x)。合并的有機層經硫酸鈉干燥,過濾并且在真空中濃縮,以得到為灰白色固體的標題化合物(9mg,73%產率)。ms?m/z:149.1[m+h]+,esi?pos。
183、中間體b4:4.5-二甲基異噻唑-3-胺
184、
185、步驟1:n-(5-甲基異噻唑-3-基)氨基甲酸叔丁酯
186、
187、在-10℃下,向5-甲基異噻唑-3-甲酸(300mg,2.1mmol)和三乙胺(233mg,322ul,2.31mmol)在四氫呋喃(9ml)的溶液中滴加氯甲酸乙酯(250mg,220ul,2.31mmol)。將混合物在-10℃下攪拌30min。然后緩慢添加疊氮化鈉(682mg,10.5mmol)在水(3ml)中的溶液,允許混合物溫熱至室溫并攪拌1h。將混合物用冰/水(20ml)稀釋并用二氯甲烷(3x?20ml)萃取。合并的有機層經硫酸鈉干燥,過濾并且在真空中濃縮。將殘余物溶解在甲苯(9ml)中,添加叔丁醇(4.5ml)并將混合物加熱至80℃持續3.5h。將混合物在真空中濃縮。將殘余物通過快速柱色譜法(硅膠,庚烷中的0-30%乙酸乙酯)純化,以得到為白色固體的n-(5-甲基異噻唑-3-基)氨基甲酸叔丁酯(233mg,52%產率)。ms?m/z:159.0[m-異丁烯+h]+,esi?pos。
188、步驟2:n-(4-溴-5-甲基-異噻唑-3-基)氨基甲酸叔丁酯
189、
190、在22℃下,向n-(5-甲基異噻唑-3-基)氨基甲酸叔丁酯(203mg,0.947mmol)在乙酸(2.6ml)中的溶液添加乙酸鈉(256mg,3.13mmol),隨后添加溴(332mg,108ul,2.08mmol)。將管密封并將混合物在50℃下攪拌1.5h。冷卻后,將混合物倒入冷的10%碳酸鈉水溶液(60ml)中。將水層用乙酸乙酯(2x?50ml)萃取,然后將合并的有機層經硫酸鈉干燥,過濾并且在真空中濃縮。
191、將殘余物通過快速柱色譜法(硅膠,庚烷中的0-30%乙酸乙酯)純化,以得到為淺黃色油狀物的n-(4-溴-5-甲基-異噻唑-3-基)氨基甲酸叔丁酯(128mg,46%產率)。ms?m/z:237.0,239.0[m+h]+,esi?pos。
192、步驟3:n-(4.5-二甲基異噻唑-3-基)氨基甲酸叔丁酯
193、
194、在22℃下,向n-(4-溴-5-甲基-異噻唑-3-基)氨基甲酸叔丁酯(63mg,0.215mmol)在1,4-二噁烷(1.3ml)中的溶液中添加三甲基環硼氧烷(67mg,75ul,0.537mmol),隨后添加在水(0.26ml)中的碳酸鉀(119mg,0.86mmol)。將混合物通過用氬氣吹掃10min脫氣。然后添加1,1'-雙(二苯基膦基)二茂鐵-二氯化鈀(ii)二氯甲烷絡合物(8.8mg,0.011mmol)。用氬氣吹掃后,將管密封并加熱至100℃持續1.5h。將混合物冷卻至室溫,并進一步添加三甲基環硼氧烷(67mg,0.537mmol),并在100℃下繼續加熱一小時。將殘余物用飽和碳酸氫鈉水溶液(10ml)處理并用乙酸乙酯(2x?10ml)萃取。合并的有機層經硫酸鈉干燥,過濾并且在真空中濃縮,并且將殘余物通過快速柱色譜法(硅膠,庚烷中的0-100%乙酸乙酯)純化,以得到為白色固體的n-(4,5-二甲基異噻唑-3-基)氨基甲酸叔丁酯(20mg,40%)。ms?m/z:173.0[m-異丁烯+h]+,esi?pos。
195、步驟4:4.5-二甲基異噻唑-3-胺
196、
197、在22℃下,向n-(4,5-二甲基異噻唑-3-基)氨基甲酸叔丁酯(24mg,0.105mmol)在二氯甲烷(1ml)中的溶液中添加三氟乙酸(120mg,80ul,1.05mmol)。將混合物在22℃下攪拌18h,然后在真空中濃縮。將殘余物用飽和碳酸氫鈉水溶液(10ml)處理并用乙酸乙酯(2x10ml)萃取。合并的有機層經硫酸鈉干燥,過濾并且在真空中濃縮,以得到為淺黃色固體的標題化合物(12.5mg,93%產率)。ms?m/z:129.0[m+h]+,esi?pos。
198、中間體b5:5-乙基-4-甲基-異噁唑-3-胺
199、
200、步驟1:n-(5-乙基-4-甲基-異噁唑-3-基)氨基甲酸叔丁酯
201、
202、將5-乙基-4-甲基-異噁唑-3-甲酸(cas1119452-16-2,208mg,1.31mmol)溶解在叔丁醇(2.9ml)中。添加三乙胺(266mg,366ul,2.63mmol),隨后添加疊氮磷酸二苯酯(398mg,311ul,1.45mmol),并將所得溶液在90℃下加熱18h。將溶劑在減壓下去除。將所得材料在乙酸乙酯與水之間分配。將層萃取,并且將水層用乙酸乙酯萃取兩次。合并的有機層經硫酸鈉干燥,過濾并在減壓下濃縮。將殘余物通過快速色譜法(硅膠,庚烷中的0%至100%乙酸乙酯)純化,以得到為白色粉末的n-(5-乙基-4-甲基-異噁唑-3-基)氨基甲酸叔甲基酯(100mg,33%)。ms?m/z:227.1[m+h]+,esi?pos。
203、步驟2:5-乙基-4-甲基-異噁唑-3-胺
204、
205、將n-(5-乙基-4-甲基-異噁唑-3-基)氨基甲酸叔丁酯(100mg,0.433mmol)溶解在超干燥的1,2-二氯乙烷(1ml)中,并且將三氟乙酸(248.64mg,168ul,2.18mmol)小心滴加到混合物中。將所得溶液在室溫下攪拌20h。完成后,將飽和碳酸氫鈉溶液添加到混合物,并用二氯甲烷萃取兩次。合并的有機層經硫酸鈉干燥,過濾并且在減壓下濃縮,以得到為黃色油狀物的標題化合物(47mg,81%),其不經進一步純化即使用。ms?m/z:127.1[m+h]+,esipos。
206、中間體b6:4-氯-5-乙基-異噻唑-3-胺
207、
208、類似于中間體b1,由4,4,5,5-四甲基-2-乙烯基-1,3,2-二氧雜環戊硼烷代替4,4,5,5-四甲基-2-(丙-1-烯-1-基)-1,3,2-二氧雜環戊硼烷來制備為白色固體標題化合物,ms(esi)m/z:163.0[m+h]+。
209、中間體b7:4-乙基-5-甲基-異噻唑-3-胺
210、
211、步驟1:5-甲基-4-乙烯基-異噻唑-3-胺
212、
213、在氬氣下添加n-(4-溴-5-甲基-異噻唑-3-基)氨基甲酸叔丁酯(參見中間體b4步驟2,300mg,0.972mmol)、4,4,5,5-四甲基-2-乙烯基-1,3,2-二氧雜環戊硼烷(195mg,1.26mmol)、1,4-二噁烷(4ml)、2m碳酸鈉水溶液(1.46ml,2.92mmol)和1,1'-雙(二苯基膦基)二茂鐵-二氯化鈀(ii)二氯甲烷絡合物(80mg,0.097mmol)。將反應混合物在90℃攪拌過夜。冷卻至室溫后,將反應混合物倒入水中并用乙酸乙酯萃取兩次。將合并的有機層用鹽水洗滌,經硫酸鈉干燥,過濾并且在真空中濃縮。將殘余物溶解在二氯甲烷(2ml)中,添加三氟乙酸(1.11g,749ul,9.72mmol),并將反應混合物在室溫下攪拌18h。將反應混合物倒入飽和碳酸氫鈉溶液中并用乙酸乙酯萃取兩次。將合并的有機層用鹽水洗滌,經硫酸鈉干燥,過濾并且在真空中濃縮。將粗制材料通過快速色譜法(硅膠,庚烷中的0%至50%乙酸乙酯)純化,以得到為灰白色固體的5-甲基-4-乙烯基-異噻唑-3-胺(76mg,53%產率)。ms(esi)m/z:141.1[m+h]+。
214、步驟2:4-乙基-5-甲基-異噻唑-3-胺
215、
216、在氬氣下,向(5-甲基-4-乙烯基-異噻唑-3-基)胺(76mg,0.542mmol)在乙酸乙酯(20ml)中的溶液中添加活性炭載鈀(58mg,0.054mmol)。將反應物置于氫氣氣氛下并將黑色懸浮液在室溫下攪拌90min。將反應混合物通過硅藻土墊過濾并用乙酸乙酯洗滌。在真空中濃縮濾液,以得到為灰白色半固體的標題化合物(66mg,86%產率)。ms(esi)m/z:143.1[m+h]+。
217、中間體b8:4-氯-5-甲氧基-異噻唑-3-胺
218、
219、步驟1:4-氯-5-甲氧基-異噻唑-3-甲酸
220、
221、將氫化鈉(60%分散在礦物油中,3.03g,75.75mmol)小心地添加至甲醇(105ml,2.59mol)并將混合物在25℃下攪拌30min。接下來,添加4,5-二氯異噻唑-3-甲酸(cas131947-13-2,3.0g,15.15mmol)并將混合物在80℃下攪拌6h。添加水(5ml)并通過添加1n鹽酸將混合物的ph調節至<7。混合物用乙酸乙酯(3x?200ml)萃取,并且合并的有機層經硫酸鈉干燥,過濾并且在減壓下濃縮。殘余物通過制備型hplc(柱:phenomenex?luna?c18(250*70mm,10um),水-乙腈)純化,以得到為灰白色固體的4-氯-5-甲氧基-異噻唑-3-甲酸(1.0g,34%產率)。1h?nmr(400mhz,dmso-d6)δ=4.16(s,3h)。
222、步驟2:4-氯-5-甲氧基-異噻唑-3-胺
223、
224、向4-氯-5-甲氧基-異噻唑-3-甲酸(300mg,1.55mmol)在叔丁醇(10ml)中的溶液中添加三甲胺(188mg,1.86mmol)和疊氮磷酸二苯酯(415mg,1.7mmol)。將混合物在90℃下攪拌12h。lcms顯示叔丁基基團已經裂解。將反應混合物減壓濃縮。殘余物通過制備型hplc(柱:phenomenex?luna?c18?150*25mm*10um,水-乙腈)純化,以得到為灰白色固體的標題化合物(120mg,30%產率)。ms(esi)m/z:164.9[m+h]+。中間體b9:4-氯-5-乙氧基-異噻唑-3-胺
225、
226、類似于中間體b8,由乙醇代替甲醇來制備標題化合物。白色固體,ms(esi)m/z:151.1[m-et+h]+。179.1[m+h]+。
227、中間體b10:3-(3-氨基-4-氯-異噻唑-5-基)丙腈三氟乙酸酯
228、
229、步驟1:4-氯-5-(2-氰基乙基)異噻唑-3-甲酸甲酯
230、
231、在反應管中加入4,5-二氯異噻唑-3-甲酸甲酯(cas166668-76-4,1.15g,5.42mmol)和tbuxphos?pd?g3(862mg,1.08mmol)。將管密封、脫氣并填充四氫呋喃(超干燥,40ml)和2-氰乙基溴化鋅溶液(0.5m的thf,16.3ml,8.13mmol)。將所得混合物在60℃加熱過夜。將反應混合物濃縮并通過快速色譜法(硅膠,庚烷中的0-70%乙酸乙酯)純化,以得到為黃色油狀物的4-氯-5-(2-氰基乙基)異噻唑-3-甲酸甲酯(209mg,17%),ms(esi)m/z:231.1[m+h]+,esi?pos。
232、步驟2:4-氯-5-(2-氰基乙基)異噻唑-3-甲酸
233、
234、向4-氯-5-(2-氰基乙基)異噻唑-3-甲酸甲酯(50mg,0.173mmol)在四氫呋喃(1ml)中的溶液中添加氫氧化鋰溶液(1m水,(260ul,0.26mmol),并將反應混合物在室溫下攪拌30min。將反應混合物用鹽酸(1m水)酸化并用乙酸乙酯萃取兩次。合并的有機層用鹽水洗滌,經硫酸鈉干燥,過濾并濃縮,以得到為淺棕色油狀物的4-氯-5-(2-氰基乙基)異噻唑-3-甲酸(50mg,99%)。ms(esi)m/z:217.1[m+h]+,esi?pos。
235、步驟3:n-[4-氯-5-(2-氰基乙基)異噻唑-3-基]氨基甲酸叔丁酯
236、
237、向4-氯-5-(2-氰基乙基)異噻唑-3-甲酸(50mg,0.173mmol)在叔丁醇(1ml)中的溶液中添加疊氮磷酸二苯酯(52mg,41ul,0.190mmol)和三乙胺(35mg,48ul,0.346mmol),并將反應混合物在90℃下攪拌2h。將反應混合物用水稀釋并用乙酸乙酯萃取兩次。將合并的有機層用鹽水洗滌,經硫酸鈉干燥,過濾并且濃縮至干。將殘余物通過快速色譜法(硅膠,庚烷中的0-100%乙酸乙酯)純化,以得到為無色油狀物的n-[4-氯-5-(2-氰基乙基)異噻唑-3-基]氨基甲酸叔丁酯(19mg,38%產率)。ms(esi)m/z:286.1[m-h]-,esi?neg。
238、步驟4:3-(3-氨基-4-氯-異噻唑-5-基)丙腈三氟乙酸酯
239、
240、向n-[4-氯-5-(2-氰基乙基)異噻唑-3-基]氨基甲酸叔丁酯(19mg,0.066mmol)在二氯甲烷(0.5ml)中的攪拌溶液中添加三氟乙酸(75mg,51ul,0.66mmol)并將反應混合物在室溫下攪拌30min。將反應混合物在真空中濃縮,以得到為無色半固體的標題化合物(20mg,100%)。ms(esi)m/z:188.0[m+h]+,esi?pos。
241、中間體b11:5-甲基異噻唑-3-胺
242、
243、中間體b11是商業的(cas128146-85-0)。
244、中間體b12:4-環丙基-5-甲基-異噻唑-3-胺
245、
246、在室溫下向n-(4-溴-5-甲基-異噻唑-3-基)氨基甲酸叔丁酯(參見中間體b4步驟2,200mg,0.682mmol)在甲苯(4ml)中的溶液中添加環丙基硼酸(147mg,1.71mmol),隨后添加在水(0.8ml)中的磷酸鉀(579mg,2.73mmol)。將混合物通過用氬氣吹掃10min脫氣。然后添加乙酸鈀(ii)(7.5mg,0.034mmol),隨后添加三環己基膦(19mg,0.068mmol),并將混合物再次用氬氣吹掃。將管密封并加熱至100℃持續3h,然后將混合物冷卻至22℃,用碳酸鈉水溶液(30ml)處理并用乙酸乙酯(2x?30ml)萃取。合并的有機層經硫酸鈉干燥,過濾并且在真空中濃縮。將殘余物通過快速柱色譜法(硅膠,庚烷中的0-100%乙酸乙酯)純化,以得到n-(4-環丙基-5-甲基-異噻唑-3-基)氨基甲酸叔丁酯(45mg,26%產率)和4-環丙基-5-甲基-異噻唑-3-胺(51mg,48%產率)。
247、將n-(4-環丙基-5-甲基-異噻唑-3-基)氨基甲酸叔丁酯(36mg,0.142mmol)溶解在二氯甲烷(1.3ml)中,并且在室溫下添加三氟乙酸(162mg,108ul,1.42mmol)并將混合物攪拌過夜,以獲得第二批標題化合物。將混合物在真空中濃縮并將殘余物用飽和碳酸氫鈉水溶液(10ml)處理并用乙酸乙酯(2x?10ml)萃取。將有機層經硫酸鈉干燥,過濾并且在真空中濃縮。將殘留物通過快速柱色譜法(硅膠,庚烷中的10-70%乙酸乙酯)純化,以得到為灰白色固體的標題化合物(18mg,80%產率)。ms(esi)m/z:155.0[m+h]+,esi?pos。
248、中間體b13:3-氨基-5-甲基-異噻唑-4-甲腈
249、
250、中間體b13是已知化合物(cas13599-06-9),其例如由hartke,k.等人在archivder?pharmazie?und?berichte?der?deutschen?pharmazeutischen?gesellschaft(1968),301(8),611-21中描述。
251、中間體b14:4-甲氧基-5-甲基-異噻唑-3-胺
252、
253、步驟1:4-碘-5-甲基-異噻唑-3-甲酸
254、
255、向5-甲基異噻唑-3-甲酸(cas110632-59-2,500mg,3.49mmol)在三氟乙酸(10ml)中的攪拌溶液中添加n-碘代琥珀酰亞胺(1.1g,4.89mmol),并且將反應混合物在20℃下攪拌18h。添加水并將混合物用乙酸乙酯萃取兩次。將合并的有機物用水洗滌,經硫酸鈉干燥,過濾并且在真空中濃縮,以獲得為灰白色固體的4-碘-5-甲基-異噻唑-3-甲酸(383mg,41%產率),其不經進一步純化即使用。ms(esi)m/z:269.8[m+h]+,esi?pos。
256、步驟2:4-甲氧基-5-甲基-異噻唑-3-甲酸
257、
258、向甲醇鈉(558mg,10.3mmol)的甲醇(50ml)溶液中添加4-碘-5-甲基-異噻唑-3-甲酸(556mg,2.1mmol)和碘化亞銅(i)(118mg,0.62mmol)。將反應混合物在80℃下攪拌48h,然后用水稀釋,蒸發并用1m磷酸酸化至ph?2-3。將水層用乙酸乙酯萃取(3x)并通過制備型hplc(柱:phenomenex?luna?c18(250*70mm,10um),水-乙腈)純化,以獲得為灰白色固體的4-甲氧基-5-甲基-異噻唑-3-甲酸(110mg,30%產率)。ms(esi)m/z:174.0[m+h]+,esi?pos。
259、步驟3:4-甲氧基-5-甲基-異噻唑-3-胺
260、
261、將4-甲氧基-5-甲基-異噻唑-3-甲酸(100mg,0.58mmol)、疊氮磷酸二苯酯(0.12ml,0.58mmol)和三乙胺(0.16ml,1.16mmol)在叔丁醇(1.0ml)中混合并在90℃下加熱18h。將反應混合物用碳酸鉀溶液淬滅并用乙酸乙酯萃取并蒸發。將殘余物溶解在二氯甲烷(0.5ml)中,添加三氟乙酸(0.5ml)并將反應混合物在20℃下攪拌18h。將混合物用稀釋的氫氧化銨水溶液淬滅并用乙酸乙酯萃取兩次。合并的有機層經硫酸鈉干燥,過濾并且蒸發,以獲得為無色油狀物的標題化合物(22mg,26%產率)。ms(esi)m/z:145.0[m+h]+,esi?pos。
262、中間體b15:4-甲氧基-5-丙基-異噻唑-3-胺
263、
264、步驟1:5-溴-4-甲氧基-異噻唑-3-甲酸乙酯
265、
266、在室溫下,將碘甲烷(676mg,4.76mmol)添加至5-溴-4-羥基-異噻唑-3-甲酸乙酯(cas?321601-49-4,描述于wo2001014339中,400mg,1.59mmol)和碳酸鉀(658mg,4.76mmol)在二甲基甲酰胺中的溶液,并將反應混合物在室溫下攪拌18h。添加水后,將混合物用乙酸乙酯萃取兩次。合并的有機相用水和飽和氯化鈉水溶液洗滌。經硫酸鈉干燥并去除溶劑,以得到5-溴-4-甲氧基-異噻唑-3-甲酸乙酯(270mg,90%純度,58%產率)。ms(esi)m/z:266.0,268.0[m+h]+,esi?pos。
267、步驟2:4-甲氧基-5-丙-1-烯基-異噻唑-3-甲酸乙酯
268、
269、向5-溴-4-甲氧基-異噻唑-3-甲酸乙酯(206mg,0.77mmol)和4,4,5,5-四甲基-2-(丙-1-烯-1-基)-1,3,2-二氧雜環戊硼烷(156mg,0.93mmol)在1,4-二噁烷(1ml)和水(0.25ml)中的的混合物中添加1,1'-雙(二苯基膦基)二茂鐵-二氯化鈀(ii)二氯甲烷絡合物(28.5mg,0.03mmol)和碳酸銫(507mg,1.56mmol)。將反應混合物抽真空并用氬氣重新填充3次,并在90℃下攪拌1.5h。然后將混合物冷卻并用水(20ml)稀釋并用乙酸乙酯(80ml)洗滌。將合并的有機層用鹽水(40ml)洗滌,經硫酸鈉干燥,過濾并且在減壓下濃縮,以得到4-甲氧基-5-丙-1-烯基-異噻唑-3-甲酸乙酯(170mg,84%產率),ms(esi)m/z:228.0[m+h]+,esi?pos。
270、步驟3:4-甲氧基-5-丙基-異噻唑-3-甲酸乙酯
271、
272、向4-甲氧基-5-[(e)-丙-1-烯基]異噻唑-3-甲酸乙酯(170mg,0.75mmol)在甲醇(10ml)中的溶液中添加20℃下是炭載鈀(80mg)。將反應混合物在氫氣的氣氛和5bar下攪拌18h。將反應混合物過濾并且在減壓下蒸發溶劑,以得到4-甲氧基-5-丙基-異噻唑-3-甲酸乙酯(168mg,85%產率),ms(esi)m/z:230.0[m+h]+,esi?pos。
273、步驟4:4-甲氧基-5-丙基-異噻唑-3-甲酸
274、
275、將4-甲氧基-5-丙基-異噻唑-3-甲酸乙酯(151mg,0.66mmol)和氫氧化鈉(40mg,0.99mmol)在乙醇(1.5ml)和水(0.5ml)中混合在一起,并在20℃下攪拌18h。接下來將其蒸發并用二氯甲烷(3x?10ml)萃取。將有機層丟棄。將水相用磷酸酸化至ph?4,并用乙酸乙酯(3×20ml)萃取。將有機萃取物用鹽水(5×20ml)洗滌,經硫酸鈉干燥,并在減壓下濃縮,以得到為固體的4-甲氧基-5-丙基-異噻唑-3-甲酸(110mg,83%產率),ms(esi)m/z:202.2[m+h]+,esi?pos。
276、步驟5:n-(4-甲氧基-5-丙基-異噻唑-3-基)氨基甲酸叔丁酯
277、
278、4-甲氧基-5-丙基-異噻唑-3-甲酸(110mg,0.55mmol)、二苯基膦疊氮化物(150mg,0.12ml,0.55mmol)和三乙胺(111mg,0.15ml,1.09mmol)在叔丁醇(0.5ml)中的溶液在90℃下加熱18h。將反應混合物用碳酸鉀溶液淬滅,并用乙酸乙酯萃取,以獲得n-(4-甲氧基-5-丙基-異噻唑-3-基)氨基甲酸叔丁酯(118mg,53%產率),ms(esi)m/z:217.02[m-tbu+h]+,esi?pos。
279、步驟6:4-甲氧基-5-丙基-異噻唑-3-胺
280、
281、向n-(4-甲氧基-5-丙基-異噻唑-3-基)氨基甲酸叔丁酯(118mg,0.43mmol)在二氯甲烷(0.5ml)中的攪拌溶液中添加三氟乙酸(0.5ml),并且將反應混合物在20℃下攪拌18h。添加水和氫氧化銨溶液(25%)并將混合物用乙酸乙酯萃取。將合并的有機層用水洗滌,經硫酸鈉干燥,過濾并且在真空中濃縮,以獲得為油狀物的標題化合物(87mg,85%純度,99%產率),ms(esi)m/z:173.0[m+h]+,esi?pos。
282、中間體b16:4-甲基-5-丙基-異噁唑-3-胺
283、
284、中間體b16是商業的(cas1207175-25-4)。
285、中間體b17:4.5-二甲基異噁唑-3-胺
286、
287、中間體b17是商業的(cas13999-39-8)。
288、中間體b18:4-氯-5-甲基-異噁唑-3-胺
289、
290、中間體b18是商業的(cas?5819-39-6)。
291、中間體b19:4-溴-5-甲基-異噁唑-3-胺
292、
293、中間體b19是商業的(cas?5819-40-9)。
294、實例
295、實例1:6-氯-n-(4-氯-5-丙基-異噻唑-3-基)-1h-吲哚-3-磺酰胺
296、
297、向4-氯-5-丙基-異噻唑-3-胺鹽酸鹽(中間體b1,20.5mg,0.082mmol)在超干燥的二氯甲烷(0.5ml)中的溶液中添加二異丙基乙胺(32mg,43ul,0.245mmol),隨后添加6-氯-1h-吲哚-3-磺酰氯(中間體a1,26.5mg,0.106mmol)。將反應混合物在23℃攪拌2h,然后用水稀釋并用二氯甲烷(2x?7ml)萃取。將合并的有機層用鹽水洗滌,經硫酸鈉干燥,過濾并且濃縮至干。將粗制化合物通過制備型hplc(柱:ymc-triart?c18,12nm,5um,100x?30mm,乙腈/水+0.1%?hcooh)純化,以得到為白色泡沫的標題化合物(22mg,66%產率)。ms?m/z:390.1[m+h]+,esi?pos。
298、類似于實例1,通過偶聯指定的磺酰氯中間體a和胺中間體b制備以下實例2-4。
299、
300、實例5:6-氯-n-(4-氯-5-甲基-異噻唑-3-基)-1h-吲哚-3-磺酰胺
301、
302、向4-氯-5-甲基-異噻唑-3-胺(中間體b3,26mg,0.166mmol)在超干燥的二氯甲烷(0.6ml)中的溶液中添加二異丙基乙胺(43mg,58ul,0.332mmol)和6-氯-1h-吲哚-3-磺酰氯(中間體a1,54mg,0.216mmol),并將混合物在室溫下攪拌45min。然后將其用乙酸乙酯和水稀釋,并將水相用乙酸乙酯萃取兩次。合并的有機層經硫酸鈉干燥,過濾并且在真空中濃縮。將殘余物通過快速色譜法(硅膠,庚烷中的0-50%乙酸乙酯)純化,以得到為白色固體的標題化合物(21mg,35%)。ms?m/z:362.2[m+h]+,esi?pos。
303、類似于實例5,通過偶聯指定的磺酰氯中間體a和胺中間體b制備以下實例6-14。
304、
305、
306、
307、實例15:6-氯-n-(4-氰基-5-甲基-異噻唑-3-基)-1h-吲哚-3-磺酰胺
308、
309、向3-氨基-5-甲基-異噻唑-4-甲腈(中間體b13,25mg,0.180mmol)和6-氯-1h-吲哚-3-磺酰氯(中間體a1,57mg,0.216mmol)的混合物添加吡啶(800ul)。將反應混合物在室溫攪拌1h,然后將溶液在真空中濃縮。將粗制材料通過快速色譜法(硅膠,庚烷中的0-60%乙酸乙酯)純化,隨后通過制備型hplc(柱:ymc-triart?c18,12nm,5um,100x30mm,乙腈/水+0.1%?hcooh)純化,以得到為白色固體的標題化合物(12mg,19%產率)。ms(esi)m/z:353.0[m+h]+。
310、類似于實例15,通過偶聯指定的磺酰氯中間體a和胺中間體b制備以下實例16-17。
311、
312、
313、實例18:6-氯-n-(4-甲基-5-丙基-異噁唑-3-基)-1h-吲哚-3-磺酰胺
314、
315、向4-甲基-5-丙基-異噁唑-3-胺(中間體b16,38mg,0.27mmol)和n,n-二異丙基乙胺(52mg,0.07ml,0.41mmol)在干燥吡啶(1ml)中的溶液中添加6-氯-1h-吲哚-3-磺酰氯(中間體a1,75mg,0.3mmol),并將混合物在室溫下攪拌18h。然后將混合物通過制備型hplc(柱:ymc?triart?c18,100x20mm,5um,水/乙腈+0.1%?nh4oh)純化,以得到為黃色固體的標題化合物(8mg,8%產率)。ms(esi)m/z:354.0[m+h]+。實例19:6-氯-n-(4,5-二甲基異噁唑-3-基)-1h-吲哚-3-磺酰胺
316、
317、類似于實例18,由中間體b17代替中間體b16來制備標題化合物。橙色固體,ms(esi)m/z:326.0[m+h]+。
318、實例20:6-氯-n-(4-氯-5-甲基-異噁唑-3-基)-1h-吲哚-3-磺酰胺
319、
320、4-氯-5-甲基-異噁唑-3-胺(中間體b18,40mg,0.30mmol)、6-氯-1h-吲哚-3-磺酰氯(中間體a1,50mg,0.20mmol)和4-二甲基氨基吡啶(2.5mg,0.02mmol)被引入反應瓶中。在室溫下添加吡啶(超干燥,3ml)并將混合物在100℃下攪拌1h。將反應混合物冷卻至室溫,用檸檬酸(1m水)稀釋并用乙酸乙酯萃取兩次。將合并的有機層用水和鹽水洗滌,經硫酸鈉干燥并濃縮至干。殘余物通過制備型hplc(柱:ymc-triart?c18,12nm,5um,100x?30mm,乙腈/水+0.1%?hcooh)純化,以得到為白色固體的標題化合物(19mg,27%產率),ms(esi)m/z:346.1[m+h]+。
321、實例21:n-(4-溴-5-甲基-異噁唑-3-基)-6-氯-1h-吲哚-3-磺酰胺
322、
323、類似于實例20,由中間體b19代替中間體b18來制備標題化合物。白色固體,ms(esi)m/z:391.9[m+h]+。
324、實例a
325、式i化合物可以以本身已知的方式用作生產以下組成的片劑的活性成分:
326、
327、實例b
328、式i化合物可以以本身已知的方式用作生產以下組成的膠囊的活性成分:
329、
330、
- 還沒有人留言評論。精彩留言會獲得點贊!