一種阿苯達唑及其代謝物的檢測方法及應用與流程
本發明屬于藥物檢測
技術領域:
,具體涉及一種阿苯達唑及其代謝物的檢測方法及應用。
背景技術:
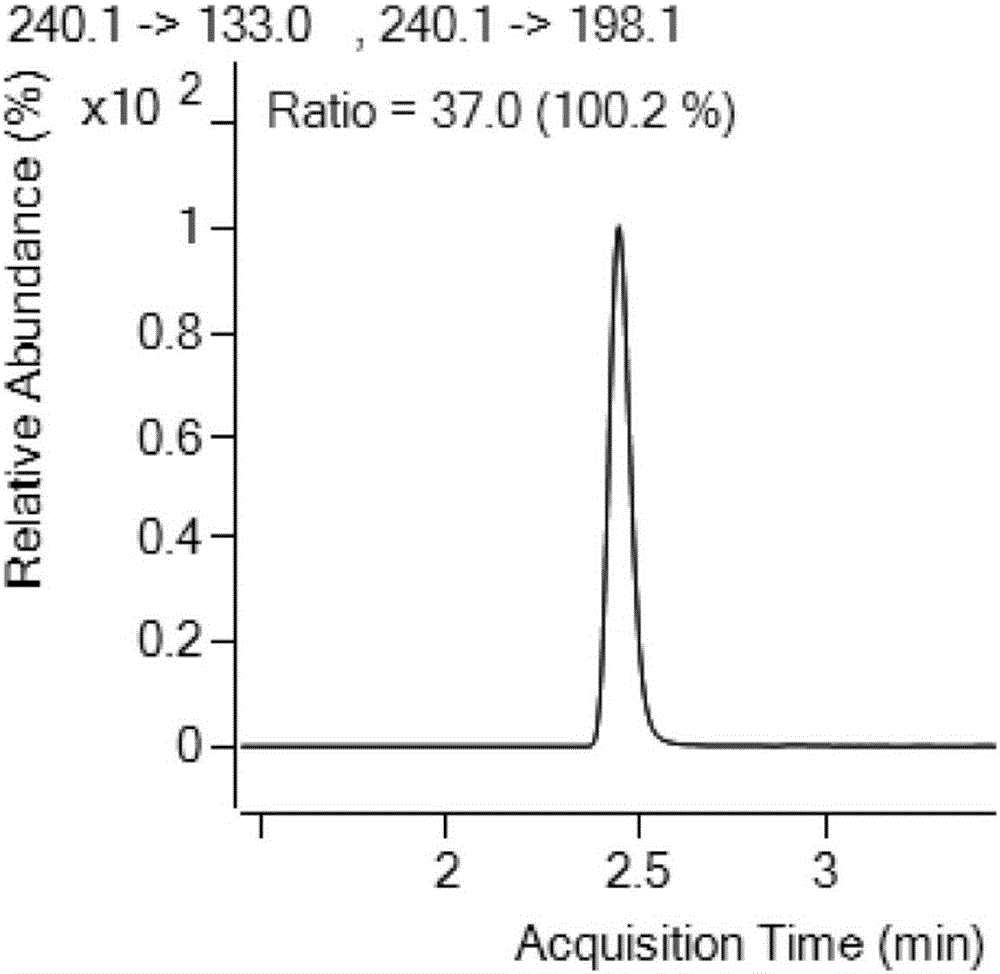
:目前,檢測阿苯達唑的方法中使用乙醚、乙腈等毒性大的溶劑,或者直接使用乙腈提取,這些方法不僅使樣品提取不充分,而且目標化合物在提取過程中極易遭到破壞,從而影響定量結果。技術實現要素:為了解決現有技術中的問題,本發明采取的技術方案是:一種阿苯達唑及其代謝物的檢測方法,利用2,6-二叔丁基對甲酚和NaOH提取樣品中的阿苯達唑及其代謝物,之后使用高效液相色譜質譜聯用儀檢測;所述2,6-二叔丁基對甲酚的濃度為1%;所述NaOH溶液的濃度為50%。在上述方案的基礎上,所述阿苯達唑及其代謝物的檢測方法步驟如下:1)提取純化稱取樣品5.00g,加入5mL水、150μL50%NaOH溶液和1mL1%2,6-二叔丁基對甲酚,渦旋混合,再加入20mL乙酸乙酯,振蕩1min超聲提取20min,4000r/min離心5min,取上清液,向殘渣中加入10mL乙酸乙酯重復提取一次,合并兩次上清液于梨形瓶中,40℃旋蒸至干;向旋干后的梨形瓶中加入6mL乙腈飽和正己烷,振蕩混勻,倒入離心管中,再向梨形瓶中加入2mL乙腈,渦旋超聲1min后一并倒入離心管中,向離心管中加入6mL0.1mol/L的鹽酸溶液,渦旋2min,4000r/min離心5min,除去正己烷層,備用;若乳化,超聲并用吸管攪拌后離心;2)凈化柱凈化用5mL甲醇和5mL水平衡活化PCX固相萃取柱后,取提取液過PCX凈化柱,分別用3mL水和3mL甲醇淋洗,然后用15mL體積濃度為5%氨水甲醇溶液洗脫,接收的洗脫液于40℃旋蒸至干后;分別加入1mL甲醇和1mL水,渦旋,過0.22μm尼龍濾膜,待測;3)高效液相色譜質譜聯用儀檢測質譜參數:離子源AJSESI源;GasTemp:325℃;GasFlow:10L/min;SheathGasTemp350℃;SheathGasflow:11L/min;液相參數:A為0.1%甲酸水溶液,B為甲醇。在上述方案的基礎上,所述阿苯達唑及其代謝物的檢測方法應用于檢測動物源性食品中的阿苯達唑及其代謝物。在上述方案的基礎上,所述動物源性食品為肌肉、蛋類、乳類、肝臟和腎臟。本發明的有益效果:本發明方法中用2,6-二叔丁基對甲酚和NaOH提取阿苯達唑及其代謝物,毒性較小;此外,阿苯達唑及其代謝物在水溶液中呈堿性,加入質量濃度為50%NaOH水溶液可使阿苯達唑及其代謝物成分子狀態,利于有機試劑提取;加入質量濃度為1%2,6-二叔丁基對甲酚可以保護阿苯達唑不被氧化;乙酸乙酯為弱極性的有機試劑,其加入有利于阿苯達唑的提取且雜質提取的少;乙腈飽和正己烷用來去除樣品中的油脂類雜質,加入鹽酸可使阿苯達唑成離子狀態有利于PCX凈化柱凈化;5%氨水甲醇呈強堿性,洗脫時可將PCX凈化柱吸附的阿苯達唑置換下來。本發明的方法檢測范圍較廣,相比于其他方法的單一基質,本方法不僅僅適用于動物肌肉而且也適用于肝臟等復雜基質的檢測,眾所周知,動物肝臟含有很多蛋白、脂肪、糖類、維生素和礦物質,凈化不干凈對目標化合物回收影響很大;本發明的方法使用2,6-二叔丁基對甲酚、NaOH和乙酸乙酯作為提取液,毒性小,成本低,節約時間,又能有效地減少對雜質的提取,簡化凈化步驟;本方法采用安捷倫PCX固相萃取小柱,也可用國產PCX小柱代替,有效降低檢測成本;采用高效液相色譜-串聯質譜法進行檢測,相比于單一的高效液相色譜,具有靈敏度高,重復性好,檢測限低的優勢。附圖說明圖1阿苯達唑-2-氨基砜保留時間的測定,其中縱坐標為響應,橫坐標為保留時間。圖2阿苯達唑-2-氨基砜定量定性離子明細,其中縱坐標為響應,橫坐標為保留時間。圖3阿苯達唑-2-氨基砜質譜圖,其中縱坐標為響應,橫坐標為m/z。圖4阿苯達唑亞砜保留時間的測定,其中縱坐標為響應,橫坐標為保留時間。圖5阿苯達唑亞砜定量定性離子明細,其中縱坐標為響應,橫坐標為保留時間。圖6阿苯達唑亞砜質譜圖,其中縱坐標為響應,橫坐標為m/z。圖7阿苯達唑砜保留時間的測定,其中縱坐標為響應,橫坐標為保留時間。圖8阿苯達唑砜定量定性離子明細,其中縱坐標為響應,橫坐標為保留時間。圖9阿苯達唑砜質譜圖,其中縱坐標為響應,橫坐標為m/z。圖10阿苯達唑保留時間的測定,其中縱坐標為響應,橫坐標為保留時間。圖11阿苯達唑定量定性離子明細,其中縱坐標為響應,橫坐標為保留時間。圖12阿苯達唑質譜圖,其中縱坐標為響應,橫坐標為m/z。圖13阿苯達唑-2-氨基砜的標準曲線,其中縱坐標為峰面積響應,橫坐標為阿苯達唑-2-氨基砜濃度。圖14阿苯達唑亞砜的標準曲線,其中縱坐標為峰面積響應,橫坐標為阿苯達唑亞砜濃度。圖15阿苯達唑砜的標準曲線,其中縱坐標為峰面積響應,橫坐標為阿苯達唑砜濃度。圖16阿苯達唑的標準曲線,其中縱坐標為峰面積響應,橫坐標為阿苯達唑濃度。圖17阿苯達唑及其代謝物定量離子S/N圖,其中縱坐標為響應,橫坐標為保留時間,從上到下依次為阿苯達唑-2-氨基砜、阿苯達唑亞砜、阿苯達唑砜、阿苯達唑。具體實施方式在本發明中所使用的術語,除非有另外說明,一般具有本領域普通技術人員通常理解的含義。下面結合具體實施例,并參照數據進一步詳細的描述本發明。以下實施例只是為了舉例說明本發明,而非以任何方式限制本發明的范圍。實施例1.標準溶液的配制1.1儲備液的配制分別稱量10.00mg阿苯達唑、阿苯達唑-2-氨基砜、阿苯達唑亞砜和阿苯達唑砜的標準品,溶解到甲醇中,定容到10mL,作為1000mg/L的儲備液(置于-18℃冷藏保存)。1.2標準中間液的配制分別準確量取1mL阿苯達唑、阿苯達唑-2-氨基砜、阿苯達唑亞砜和阿苯達唑砜標準品的儲備液,溶解到甲醇中,定容到10mL,作為100mg/L的標準中間液(置于-18℃冷藏保存)。1.3標準工作曲線的配制用標準中間液配制成1、5、10、20、50、100μg/L的標液(用甲醇+水(v/v=1:1)定容),作為工作曲線用的標準溶液。2.樣品處理2.1提取純化稱取樣品5.00g,加入5mL水、150μL50%NaOH溶液和1mL1%2,6-二叔丁基對甲酚,渦旋混合,再加入20mL乙酸乙酯,振蕩1min超聲提取20min,4000r/min離心5min,取上清液,向殘渣中加入10mL乙酸乙酯重復提取一次,合并兩次上清液于梨形瓶中,40℃旋蒸至干;向旋干后的梨形瓶中加入6mL乙腈飽和正己烷,振蕩混勻,倒入離心管中,再向梨形瓶中加入2mL乙腈,渦旋超聲1min后一并倒入離心管中,向離心管中加入6mL0.1mol/L的鹽酸溶液,渦旋2min,4000r/min離心5min,除去正己烷層,備用;若乳化,超聲并用吸管攪拌后離心;2.2凈化柱凈化用5mL甲醇和5mL水平衡活化PCX固相萃取柱后,取提取液過PCX凈化柱,分別用3mL水和3mL甲醇淋洗,然后用15mL體積濃度為5%氨水甲醇溶液洗脫,接收的洗脫液于40℃旋蒸至干后;分別加入1mL甲醇和1mL水,渦旋,過0.22μm尼龍濾膜,待測;3.高效液相色譜質譜聯用儀檢測使用的高效液相色譜質譜聯用儀為AgilentLC-MSMS1290/6460;質譜參數:離子源AJSESI源;GasTemp:325℃;GasFlow:10L/min;SheathGasTemp350℃;SheathGasflow:11L/min。液相參數:A為0.1%甲酸水溶液,B為甲醇。流動相梯度如表1所示:表1流動相梯度時間甲醇濃度1.00202.001004.001004.01205.5204.結果及分析4.1質譜結果阿苯達唑及其代謝物的質譜結果如表2所示:表2質譜結果4.2專屬性4.2.1阿苯達唑-2-氨基砜的專屬性50ppb阿苯達唑-2-氨基砜通過C18色譜柱分離,確定保留時間。如圖1所示,保留時間約為2.4min,且色譜的峰形較佳,無干擾峰出現,其中橫坐標為保留時間,縱坐標為響應。阿苯達唑-2-氨基砜定量定性離子明細如圖2所示,其中縱坐標為響應,橫坐標為保留時間。如圖3所示,1ppm阿苯達唑-2-氨基砜上機,通過質譜優化得到3個離子對,縱坐標為響應,橫坐標為m/z。4.2.2阿苯達唑亞砜的專屬性50ppb阿苯達唑亞砜通過C18色譜柱分離,確定保留時間。如圖4所示,保留時間約為2.9min,且色譜的峰形較佳,無干擾峰出現,其中橫坐標為保留時間,縱坐標為響應。阿苯達唑亞砜定量定性離子明細如圖5所示,其中縱坐標為響應,橫坐標為保留時間。如圖6所示,1ppm阿苯達唑亞砜上機,通過質譜優化得到3個離子對,縱坐標為響應,橫坐標為m/z。4.2.3阿苯達唑砜的專屬性50ppb阿苯達唑砜通過C18色譜柱分離,確定保留時間。如圖7所示,保留時間約為2.9min,且色譜的峰形較佳,無干擾峰出現。其中橫坐標為保留時間,縱坐標為響應。阿苯達唑砜定量定性離子明細如圖8所示,其中縱坐標為響應,橫坐標為保留時間。如圖9所示,1ppm阿苯達唑砜上機,通過質譜優化得到3個離子對,縱坐標為響應,橫坐標為m/z。4.2.4阿苯達唑的專屬性50ppb阿苯達唑砜通過C18色譜柱分離,確定保留時間。如圖10所示,保留時間約為3.159min,且色譜的峰形較佳,無干擾峰出現,橫坐標為保留時間,縱坐標為響應。阿苯達唑定量定性離子明細如圖11所示,其中縱坐標為響應,橫坐標為保留時間。如圖12所示,1ppm阿苯達唑上機,通過質譜優化各得到3個離子對,縱坐標為響應,橫坐標為m/z。4.3標準曲線4.3.1阿苯達唑-2-氨基砜的標準曲線精密吸取阿苯達唑-2-氨基砜標準品,用50%甲醇水溶液稀釋成系列濃度為1,5,10,20,50,100ng/mL標準溶液,分別進樣測定,以峰面積響應為縱坐標,阿苯達唑-2-氨基砜濃度為橫坐標。如圖13所示,得到阿苯達唑-2-氨基砜的回歸方程為Y=1378.144404*X-594.187990,R2=0.9999,由相關系數可見,在1ng/mL~100ng/mL標準曲線線性范圍內,UPLC-MSMS法測定線性關系良好,此標準曲線可以用于準確定量。4.3.2阿苯達唑亞砜的標準曲線精密吸取阿苯達唑亞砜標準品,用50%甲醇水溶液稀釋成系列濃度為1,5,10,20,50,100ng/mL標準溶液,分別進樣測定,以峰面積響應為縱坐標,阿苯達唑亞砜濃度為橫坐標。如圖14所示,得到阿苯達唑亞砜的回歸方程為Y=519.827117*X-28.361658,R2=0.9998,由相關系數可見,在1ng/mL~100ng/mL標準曲線線性范圍內,UPLC-MSMS法測定線性關系良好,此標準曲線可以用于準確定量。4.3.3阿苯達唑砜的標準曲線精密吸取阿苯達唑砜標準品,用50%甲醇水溶液稀釋成系列濃度為1,5,10,20,50,100ng/mL標準溶液,分別進樣測定,以峰面積響應為縱坐標,阿苯達唑砜濃度為橫坐標。如圖15所示,得到阿苯達唑砜的回歸方程為Y=1384.923769*X-533.206880,R2=0.9998,由相關系數可見,在1ng/mL~100ng/mL標準曲線線性范圍內,UPLC-MSMS法測定線性關系良好,此標準曲線可以用于準確定量。4.3.4阿苯達唑精密吸取阿苯達唑標準品,用50%甲醇水溶液稀釋成系列濃度為1,5,10,20,50,100ng/mL標準溶液,分別進樣測定,以峰面積響應為縱坐標,阿苯達唑濃度為橫坐標。如圖16所示,得到阿苯達唑的回歸方程為Y=8382.358953*X-2437.446609,R2=0.9999,由相關系數可見,在1ng/mL~100ng/mL標準曲線線性范圍內,UPLC-MSMS法測定線性關系良好,此標準曲線可以用于準確定量。4.4檢出限在基質中添加相當于樣品含有1.0μg/kg的阿苯達唑及其代謝物的標準品,經前處理上機檢測,計算其信噪比(S/N)>10,如圖17所示。測得本發明方法對阿苯達唑及其代謝物的檢出限為1.0μg/kg。4.5驗證基體及回收率基體及回收率的驗證如表3所示:表3基體及回收率的驗證從市場上購買牛肉、豬肝、豬腎、從超市購買雞蛋、原料乳、鯽魚基質做加標實驗回收率70%-90%之間,說明本方法穩定性良好。5.結論通過運用UPLC-LCMSMS法對動物源基體中的阿苯達唑及其代謝物進行測定,確定使用2,6-二叔丁基對甲酚、NaOH和乙酸乙酯進行提取,正己烷純化,PCX小柱進行凈化,使阿苯達唑及其代謝物色譜峰達到基線分離,峰形良好,檢測限低,靈敏度高,方法回收穩定,本研究為動物源基質中的阿苯達唑及其代謝物殘留量的測定建立了一種可靠地分析方法。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1