一種磷酸鈣/納米銀殼層復(fù)合材料的制備方法與流程
本發(fā)明屬于材料制備技術(shù)領(lǐng)域,涉及一種磷酸鈣/納米銀殼層復(fù)合材料的制備方法。
背景技術(shù):
納米銀粒子具有體積效應(yīng)、表面效應(yīng)、量子尺寸效應(yīng)以及宏觀量子隧道效應(yīng),在催化行業(yè)、生物醫(yī)藥行業(yè)、電子工業(yè)等多方面有著廣闊的應(yīng)用前景。納米銀的性質(zhì)主要取決于它的形貌與尺寸,因而納米銀的可控制備就顯得尤為重要。復(fù)雜結(jié)構(gòu)的納米銀具有特定的形貌和較大的比表面積,在表面增強(qiáng)拉曼(sers)和催化方面有很好的應(yīng)用,但納米銀的細(xì)胞毒性限制了其在生物體內(nèi)的應(yīng)用。磷酸鈣又稱磷酸三鈣,化學(xué)式為ca3(po4)2,被認(rèn)為是與生物體骨組織成分最為相似的天然無機(jī)物,具有良好的生物相容性、生物活性和生物可降解性,在生物材料領(lǐng)域有非常好的應(yīng)用前景,磷酸鈣經(jīng)常被用作為各種材料的涂層,以此來提高材料的生物相容性。
專利cn201210057548公開了一種四氧化三鐵磷酸鈣核殼納米磁性粒子及生物礦化法的制備方法,該方法通過制備四氧化三鐵納米磁性粒子并用表面改性劑對其改性,然后加入到模擬人體體液中進(jìn)行礦化,最終得到四氧化三鐵磷酸鈣核殼納米磁性粒子,但該粒子無法對藥物釋放情況進(jìn)行勘測,且合成步驟相當(dāng)繁瑣、不經(jīng)濟(jì)環(huán)保,反應(yīng)體系中過多的反應(yīng)物也會影響材料的后續(xù)使用。
技術(shù)實(shí)現(xiàn)要素:
本發(fā)明的目的在于提供一種磷酸鈣/納米銀殼層復(fù)合材料的制備方法,合成的粒子保留了納米銀的sers活性。
實(shí)現(xiàn)上述目的的技術(shù)方案如下:
一種磷酸鈣/納米銀殼層復(fù)合材料的制備方法,具體步驟如下:
步驟1,將硝酸銀溶液加熱至沸騰后,緩慢滴加檸檬酸鈉溶液,邊滴邊攪拌至反應(yīng)完全,冷卻至室溫,得到銀膠種子;
步驟2,將銀膠種子溶于抗壞血酸溶液中,滴加硝酸銀溶液,邊滴邊攪拌,得到花狀納米銀溶液;
步驟3,按花狀納米銀與乳酸鈣的摩爾比為1:0.675~1:3.375,將花狀納米銀溶液加入到乳酸鈣溶液中,超聲分散均勻后,分多次滴加磷酸鈉溶液,超聲至反應(yīng)完全,得到納米銀/磷酸鈣殼層復(fù)合材料。
步驟1中,所述的硝酸銀和檸檬酸鈉的摩爾比為1:2。
步驟2中,所述的硝酸銀和抗壞血酸的摩爾比為1:1.4。
步驟3中,所述的磷酸鈉與乳酸鈣的濃度比例為1:1.5。
步驟3中,所述的乳酸鈣的濃度為11.25mm,磷酸鈉的濃度為7.5mm。
步驟3中,加入磷酸鈉時分兩次以上加入,每次加完后超聲10s。
本發(fā)明先利用晶種法制得短棒堆積的自組裝花狀納米銀,平均粒徑在150nm,再通過共沉淀的方法在花狀納米銀表面形成一層磷酸鈣層,并通過改變?nèi)樗徕}與花狀納米銀的摩爾比,控制在納米銀表面形成的磷酸鈣層的厚度。本發(fā)明方法簡單,原料易得,合成的磷酸鈣/納米銀殼層復(fù)合材料穩(wěn)定性較好,對于羅丹明6g和燦爛綠均有較強(qiáng)的拉曼增強(qiáng)作用,當(dāng)羅丹明6g和燦爛綠的濃度低至5×10-7m時仍有明顯的拉曼增強(qiáng)效果,可用于藥物釋放的勘測。
附圖說明
圖1是實(shí)施例1制備的花狀納米銀(a),實(shí)施例1制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag1(b),實(shí)施例2制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag2(c),實(shí)施例3制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag3(d)的掃描電鏡圖。
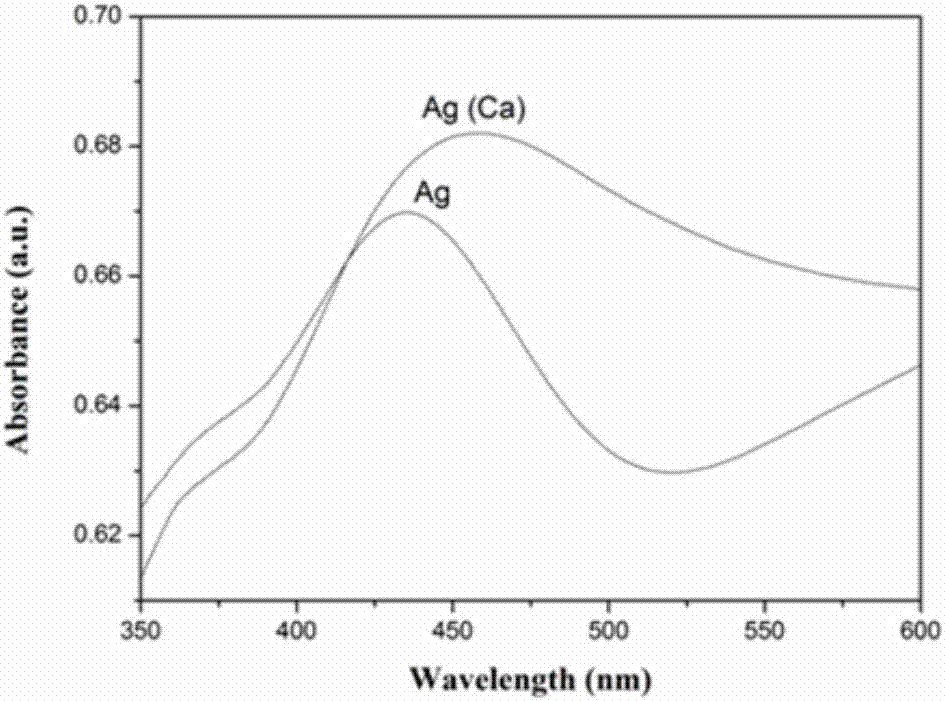
圖2是實(shí)施例1制備的花狀納米銀和磷酸鈣/納米銀殼層復(fù)合材料ca/ag1的紫外可見吸光光譜圖。
圖3是實(shí)施例1制備的花狀納米銀(a)和磷酸鈣/納米銀殼層復(fù)合材料ca/ag1(b)的xrd圖。
圖4是實(shí)施例1制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag1(a)和花狀納米銀(b)的拉曼光譜圖。
圖5是實(shí)施例1制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag1(c),實(shí)施例2制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag2(b),實(shí)施例3制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag3(a)的拉曼光譜圖。
圖6是對比例制得的磷酸鈣-納米銀復(fù)合材料ca/ag的掃描電鏡圖(a),透射電鏡圖(b)和對羅丹明6g的表面增強(qiáng)拉曼圖(c)。
具體實(shí)施方式
下面結(jié)合實(shí)施例和附圖對本發(fā)明作進(jìn)一步詳述。
實(shí)施例1
步驟1,將200ml的,濃度為2.5mm的硝酸銀溶液加熱至沸騰后,緩慢滴加10ml的,濃度為0.1m的檸檬酸鈉溶液,以滴速為30滴/分鐘邊滴加邊攪拌至反應(yīng)完全,冷卻至室溫,得到銀膠種子;
步驟2,在銀膠種子中加入35ml濃度為0.01m的抗壞血酸溶液,混勻后,邊攪拌邊加入25ml濃度為0.02m的硝酸銀溶液,得到花狀納米銀;
步驟3,在6.25ml的花狀納米銀溶液中加入1.5ml濃度為11.25mm的乳酸鈣溶液(花狀納米銀與乳酸鈣的摩爾比為1:0.675),混合均勻后,邊超聲邊滴加1.5ml濃度為7.5mm的磷酸鈉溶液,滴加完全后,得到最終產(chǎn)物ca/ag1。
實(shí)施例2
步驟1,將200ml的,濃度為2.5mm的硝酸銀溶液加熱至沸騰后,緩慢滴加10ml的,濃度為0.1m的檸檬酸鈉溶液,以滴速為30滴/分鐘邊滴加邊攪拌至反應(yīng)完全,冷卻至室溫,得到銀膠種子;
步驟2,在銀膠種子中加入35ml濃度為0.01m的抗壞血酸溶液,混勻后,邊攪拌邊加入25ml濃度為0.02m的硝酸銀溶液,得到花狀納米銀;
步驟3,在6.25ml的花狀納米銀溶液中加入3.5ml濃度為11.25mm的乳酸鈣溶液(花狀納米銀與乳酸鈣的摩爾比為1:1.575),混合均勻后,邊超聲邊滴加3.5ml濃度為7.5mm的磷酸鈉溶液,滴加完全后,得到最終產(chǎn)物ca/ag2。
實(shí)施例3
步驟1,將200ml的,濃度為2.5mm的硝酸銀溶液加熱至沸騰后,緩慢滴加10ml的,濃度為0.1m的檸檬酸鈉溶液,以滴速為30滴/分鐘邊滴加邊攪拌至反應(yīng)完全,冷卻至室溫,得到銀膠種子;
步驟2,在銀膠種子中加入35ml濃度為0.01m的抗壞血酸溶液,混勻后,邊攪拌邊加入25ml濃度為0.02m的硝酸銀溶液,得到花狀納米銀;
步驟3,在6.25ml的花狀納米銀溶液中加入7.5ml濃度為11.25mm的乳酸鈣溶液(花狀納米銀與乳酸鈣的摩爾比為1:3.375),混合均勻后,邊超聲邊滴加7.5ml濃度為7.5mm的磷酸鈉溶液,滴加完全后,得到最終產(chǎn)物ca/ag3。
圖1是實(shí)施例1制備的花狀納米銀(a),實(shí)施例1制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag1(b),實(shí)施例2制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag2(c),實(shí)施例3制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag3(d)的掃描電鏡圖。實(shí)施例1~3制得的磷酸鈣/納米銀殼層復(fù)合材料的厚度由低到高,實(shí)施例1制得的磷酸鈣/納米銀殼層復(fù)合材料的厚度最小。
從圖1a可知,通過化學(xué)還原法得到的納米銀是由棒狀堆積的花狀納米銀。
由圖1b知,花狀納米銀與乳酸鈣的摩爾比為1:0.675時,磷酸鈣殼層較薄。通過圖1b與1c的比較可知,花狀納米銀與乳酸鈣的摩爾比為(1:1.575),乳酸鈣的含量增加時,磷酸鈣殼層的厚度也隨之增大。而比較圖1c和1d時,當(dāng)花狀納米銀與乳酸鈣的摩爾比增加為(1:3.375),磷酸鈣殼層的厚度進(jìn)一步增大。因此可知,磷酸鈣殼層的厚度是與乳酸鈣與花狀納米銀的摩爾比有關(guān)系的,即殼層的厚度隨著乳酸鈣的含量的增大而增大。
圖2是實(shí)施例1制備的花狀納米銀和磷酸鈣/納米銀殼層復(fù)合材料ca/ag1的紫外可見吸光光譜圖。納米銀的吸收光譜在很大程度上是由它的粒子的尺寸來決定的。一般來說,吸收強(qiáng)度增強(qiáng)的同時也伴隨著粒子粒徑的變化。如圖2所示,納米銀吸收光譜的吸收帶在λmax=435nm處,而磷酸鈣-納米銀復(fù)合材料在它最大吸收峰的位置,相較于納米銀的吸收強(qiáng)度有明顯的增強(qiáng),同時,納米銀顆粒的粒徑的增大造成了最大吸收波長處的顯著的紅移現(xiàn)象,與圖1的掃描電鏡結(jié)果是一致的。
圖3是實(shí)施例1制備的花狀納米銀(a)和磷酸鈣/納米銀殼層復(fù)合材料ca/ag1(b)的xrd圖。如圖3a所示,x射線衍射的峰位置與銀是對應(yīng)的,峰型尖銳且無其他雜峰表明納米銀純度高且結(jié)晶性較好。由圖觀察到四個衍射峰分別位于2θ=38.17°,44.28°,69.45°,77.49°處,它們分別對應(yīng)與銀的(111),(200),(220)和(311)四個晶面,反映出納米銀是面心立方結(jié)構(gòu)。圖3b所示的磷酸鈣-納米銀復(fù)合材料的xrd峰,它的位置與納米銀相比發(fā)生了非常小程度的變化。對比納米銀,由于存在無定形磷酸鈣的殼層結(jié)構(gòu),并且其它峰的強(qiáng)度均降低,所以在2θ=21.31°位置處出現(xiàn)了一個寬峰,即磷酸鈣的峰。
圖4是實(shí)施例1制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag1(a)和花狀納米銀(b)對羅丹明6g的拉曼光譜圖。如圖4所示,羅丹明的濃度低至5×10-7m時仍可以檢測出較強(qiáng)的拉曼信號,表明合成的納米銀有較好的sers活性。
圖5是實(shí)施例1制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag1(c),實(shí)施例2制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag2(b),實(shí)施例3制備的磷酸鈣/納米銀殼層復(fù)合材料ca/ag3(a)的拉曼光譜圖。a,b,c為磷酸鈣厚度遞減的磷酸鈣-納米銀復(fù)合材料。如圖a所示,信號非常弱,吸附于殼層表面的r6g其拉曼散射強(qiáng)度是較弱的,這是由于磷酸鈣殼層過厚所造成的。比較圖c與b,圖b中峰信號較弱,但在1311,1359和1511cm-1處均出現(xiàn)模糊的峰。當(dāng)磷酸鈣殼層變薄時如圖c,出現(xiàn)較多峰,同時,幾個主要的峰都有明顯的增強(qiáng),例如:610,1131,1359和1511cm-1處的峰。因此,可知磷酸鈣-納米銀復(fù)合材料殼層的厚度與其sers活性呈負(fù)相關(guān)關(guān)系。且殼層的薄厚與反應(yīng)總體系中的ca2+濃度有關(guān)。殼層的厚度與ca2+濃度呈正相關(guān)關(guān)系,隨著ca2+濃度的增加,殼層逐漸變厚。這與之前掃描電鏡得出的結(jié)論是一致的。
對比例
步驟1,將200ml的,濃度為2.5mm的硝酸銀溶液加熱至沸騰后,緩慢滴加10ml的,濃度為0.1m的檸檬酸鈉溶液,以滴速為30滴/分鐘邊滴加邊攪拌至反應(yīng)完全,冷卻至室溫,得到銀膠種子;
步驟2,在銀膠種子中加入35ml濃度為0.01m的抗壞血酸溶液,混勻后,邊攪拌邊加入25ml濃度為0.02m的硝酸銀溶液,得到花狀納米銀;
步驟3,在6.25ml的花狀納米銀溶液中加入15ml濃度為11.25mm的乳酸鈣溶液(花狀納米銀與乳酸鈣的摩爾比為1:6.75),混合均勻后,邊超聲邊滴加15ml濃度為7.5mm的磷酸鈉溶液,滴加完全后,得到最終產(chǎn)物ca/ag。
圖6為較厚殼層厚度的磷酸鈣-納米銀復(fù)合材料的掃描電鏡圖、透射電鏡圖以及對羅丹明6g的表面增強(qiáng)拉曼圖。從6a、6b可知,通過超聲破碎法得到的磷酸鈣-納米銀復(fù)合材料在該厚度時幾乎不能完全看出花狀納米銀的花狀結(jié)構(gòu)。如圖6c所示,ca/ag對羅丹明6g的表面增強(qiáng)信號與納米銀對羅丹明6g的表面增強(qiáng)信號相比非常弱,幾乎沒有增強(qiáng)效應(yīng),這是由于磷酸鈣殼層過厚所造成的。因此,可知磷酸鈣殼層的厚度直接影響該復(fù)合材料的表面拉曼增強(qiáng)效應(yīng),本發(fā)明通過改變?nèi)樗徕}與花狀納米銀的摩爾比,控制在納米銀表面形成的磷酸鈣層的厚度,得到了生物相容性好且同時具有穩(wěn)定sers活性的復(fù)合材料,通過利用包鈣納米銀的sers活性,能夠?qū)λ幬锏尼尫胚M(jìn)行勘測。
- 還沒有人留言評論。精彩留言會獲得點(diǎn)贊!
- 一種海泡石加載磁性復(fù)合材料的制備方法及其應(yīng)用與流程
- 一種SnO2/ZnO復(fù)合微納米纖維的制備方法及其產(chǎn)品與流程
- 一種輕質(zhì)化結(jié)構(gòu)復(fù)合材料的制備方法與流程
- 一種育苗專用有機(jī)無機(jī)復(fù)合肥料及制備工藝的制造方法與工藝
- 一種復(fù)合透波材料的制備方法與流程
- 一種水性聚氨酯/超細(xì)纖維復(fù)合材料的制備方法與流程
- 一種表面改性黃麻聚乳酸復(fù)合材料的制備方法與流程
- 一種低氣味、低VOC聚乙烯復(fù)合材料及其制備裝置和方法與流程
- 一種復(fù)合單體改性導(dǎo)電材料的制備方法與流程
- 一種高導(dǎo)電復(fù)合材料的制備方法與流程
- 一種高韌性低遷移PVC納米復(fù)合材料的制備方法與流程
- 一種電磁屏蔽發(fā)泡復(fù)合材料的制備方法與流程
- 一種利用廢舊物制備復(fù)合節(jié)能材料的方法與流程
- 一種納米有機(jī)改性磷酸鋯/MC尼龍復(fù)合材料及其制備方法與流程
- 一種高強(qiáng)高韌熱固性樹脂基復(fù)合材料的制備方法及應(yīng)用與流程
- 一種晶振封裝用氧化鋯纖維?氧化釤增強(qiáng)的氧化鋁陶瓷復(fù)合材料及其制備方法與流程
- 生態(tài)砂塑復(fù)合材料組合物及制備生態(tài)砂塑復(fù)合材料的方法和所制備的材料與流程
- 一種受電弓滑板用銅/炭基復(fù)合材料的制備方法與流程
- 一種納米羥基磷灰石/氧化石墨烯復(fù)合材料的制備方法與流程
- 一種高效復(fù)合防護(hù)過濾網(wǎng)材料的制備方法與流程