一種磷氯協同苯并吡喃類阻燃劑及其制備方法與流程
本發明涉及阻燃材料技術領域,具體涉及一種磷氯協同苯并吡喃類阻燃劑及其制備方法。
背景技術:
:
阻燃劑廣泛應用于電子電器、通訊、核電、航空航天、交通、建材、室內裝飾、紡織等領域,是科學防治火災的重要措施之一。隨著社會的發展和科技的進步,阻燃劑應用領域不斷拓展,世界各國對各領域材料及制品阻燃性能要求愈加嚴格,對阻燃劑的研發與應用技術也提出了新的要求。因此,新型高效、多功能化的阻燃劑的研發具有十分重要的現實意義。
9,10-二氫-9-氧雜-10-磷酰雜菲-10-氧化物(DOPO)及其衍生物是近年來開發的新型阻燃材料,因其良好的阻燃特性和溶解性能正在受到廣泛的關注,并開展了廣泛的研究。中國專利CN101376665B公開了一種含均三嗪結構氧雜菲阻燃性化合物及其制備方法與應用;中國專利CN102757579B公開了一種含環三磷腈結構的氧雜膦菲阻燃劑及其制備方法與應用。我們課題組在中國專利ZL 201310509066.9中公開了一種磷雜菲苯并噁唑類紫外發光材料及制備方法。
苯并吡喃類化合物是一類重要的雜環化合物,廣泛應用于醫藥研發和熒光材料設計等方面,而作為阻燃材料的研究極少。特別是,以醫藥中間體6-氯-4-色滿酮為原料,制備4,6-二氯-2H-苯并吡喃-3-甲醛,再經磷雜菲功能化,從而形成磷氯協同苯并吡喃類阻燃劑化合物,并考察其相關性能的研究,在現有技術中尚未見報道。
技術實現要素:
:
本發明的目的是提供一種磷氯協同苯并吡喃類阻燃劑及其制備方法。
本發明是通過以下技術方案予以實現的:
一種式I所示的磷氯協同苯并吡喃類阻燃劑:
本發明的磷氯協同苯并吡喃類阻燃劑的合成路線如下:
本發明的磷氯協同苯并吡喃類阻燃劑的制備方法,具體包括以下步驟:
(1)摩爾比為1:1.5~3.5:1.5~3的6-氯-4-色滿酮(化合物II)、N,N-二甲基甲酰胺(DMF)及三氯氧磷(POCl3),經Vilsmeier反應合成中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III);
(2)將摩爾比為1:0.8~1.2的中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物(DOPO,化合物IV)溶于有機溶劑中,攪拌回流反應4~7小時,反應完畢后,經旋轉蒸發除去有機溶劑,以混合溶劑重結晶,真空干燥,即得目標產物(化合物I)。
步驟(1)中6-氯-4-色滿酮、N,N-二甲基甲酰胺(DMF)及三氯氧磷(POCl3)的摩爾比優選為1:2.5~3.5:1.5~3。
步驟(1)優選為在0-5℃,攪拌下緩慢滴加三氯氧磷(POCl3)到無水N,N-二甲基甲酰胺(DMF),滴加過程中控制反應溫度不超過5℃,滴加完畢后,維持0-5℃繼續攪拌反應1小時;然后,在10℃下,緩慢滴加6-氯-4-色滿酮的DMF溶液,之后,在70-90℃反應3-5小時;反應完畢后,將反應液冷卻至室溫,在快速攪拌下,緩慢傾入冰水中,靜置,抽濾、水洗、干燥,經乙醇重結晶,得到中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)。
步驟(2)中所述的有機溶劑為甲苯或二氧六環。
步驟(2)中所述的混合溶劑為甲苯-甲醇或四氫呋喃-甲醇混合溶劑。
本發明的磷氯協同苯并吡喃類阻燃劑為白色固體粉末,其熔點為226.57℃(DSC,峰值),具有良好的溶解性和熱穩定性,在241℃時失重為10%,380℃時失重為50%,在600℃時殘炭率為20.79%,790℃時殘炭率仍高達15.64%,適于用作環氧樹脂、聚酯及聚氨酯等高分子材料的阻燃劑。
本發明還保護所述的磷氯協同苯并吡喃類阻燃劑的應用,作為阻燃劑用于環氧樹脂、聚酯、聚氨酯等高分子材料。
本發明的有益效果如下:
(1)本發明通過將苯并吡喃和磷雜菲基團這兩種功能性結構單元有效結合,形成了一種新的磷雜菲功能化的磷氯協同苯并吡喃類阻燃劑,增加了阻燃劑新品種。
(2)本發明磷氯協同苯并吡喃類阻燃劑的分子結構中含有磷和氯兩種優異的阻燃元素,且阻燃元素含量高,兼具磷系和鹵系阻燃劑的優點,能充分發揮磷-氯協同和促進成炭等阻燃作用,以不同阻燃機理產生協同增效作用。高溫下,磷元素轉化為磷酸或聚磷酸促進成炭,且形成的聚磷酸膜有隔熱絕氧作用;氯元素產生氣相梯度阻燃作用,或以氣體放出形式釋放熱量,降低材料表面溫度,進而達到阻燃效果。因此,該化合物成炭性好,磷-氯協同阻燃效能高。
(3)本發明提出的制備方法,工藝簡單,操作方便,無需惰性氣體保護,易于控制,容易純化,設備投資少,所用有機溶劑均可回收利用,安全環保,適于工業化生產。
(4)本發明磷氯協同苯并吡喃類阻燃劑的分子結構中含有多個苯環、六元環結構及C-P鍵,這些結構特點使得化合物的物理化學性能穩定,能適于多種高分子材料的高溫加工;非平面性磷雜菲基團的存在,能有效抑制材料的結晶化,降低結晶度,從而使化合物具有良好的成膜性能、相容性,增加材料的可加工性能。
(5)本發明所提供的磷氯協同苯并吡喃類阻燃劑的分子結構可修飾性強,分子結構中除含有苯并吡喃和磷雜菲兩種功能基團之外,還含有羥基和較優的離去基團4-位氯原子。利用這些功能基團的反應性和磷雜菲基團的特殊功能,可以用于有機功能材料的研發。特別是可通過羥基活性基團,與高分子活性基團反應,將上述阻燃結構單元引入到高分子材料的分子結構中,從而在不影響材料物理機械性能的同時,提高高分子材料的阻燃性能及耐久性。
總之,本發明將苯并吡喃和磷雜菲基團這兩種功能性結構單元有效結合,制備方法工藝簡單,適于工業化生產,形成了一種新的磷雜菲功能化的磷氯協同苯并吡喃類阻燃劑,分子結構中同時含有磷和氯兩種阻燃元素,成炭性好,殘炭率高,磷-氯協同阻燃效能高,熱穩定性好,材料的可加工性能好,可作為阻燃劑用于環氧樹脂、聚酯及聚氨酯等高分子材料技術領域。本發明拓展了苯并吡喃類化合物的研究領域。
附圖說明:
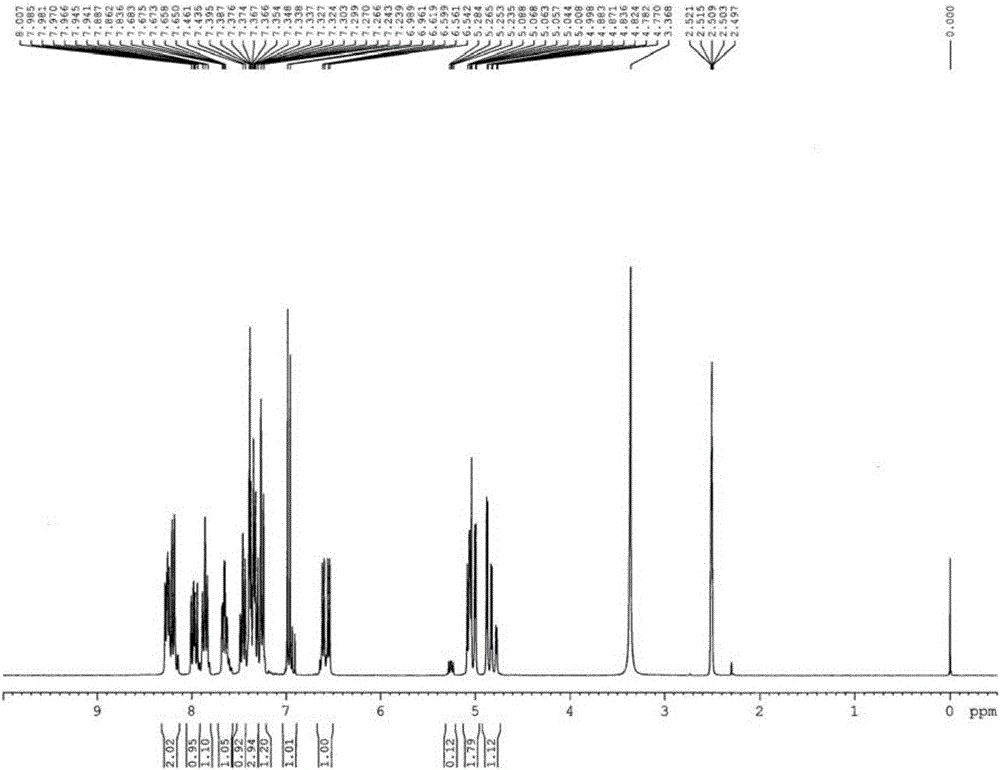
圖1是本發明的磷氯協同苯并吡喃類阻燃劑的核磁共振氫譜圖;
圖2是本發明的磷氯協同苯并吡喃類阻燃劑的紅外光譜圖;
圖3是本發明的磷氯協同苯并吡喃類阻燃劑的熱失重圖譜。
具體實施方式:
以下是對本發明的進一步說明,而不是對本發明的限制。
實驗儀器與型號:Bruker AVANCE-300核磁共振波譜儀;Nicolet Magna 760紅外光譜儀;TA DSC Q20差示掃描量熱儀;Netzsch TG 209熱重分析儀。
實施例1:磷氯協同苯并吡喃類阻燃劑的制備
(1)中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)的制備
在干燥的三口燒瓶中加入DMF(0.25mol),冰水浴冷卻至0-5℃,在攪拌下緩慢滴加POCl3(0.25mol),滴加過程中控制反應溫度不超過5℃,滴加完畢后,維持0-5℃繼續攪拌反應1小時;然后,在10℃下,緩慢滴加6-氯-4-色滿酮(化合物II)的DMF溶液(0.10mol 6-氯-4-色滿酮溶于20毫升DMF),之后,在70-90℃反應3-5小時;反應完畢后,將反應液冷卻至室溫,在快速攪拌下,緩慢傾入冰水中,靜置,抽濾、水洗、干燥,經乙醇重結晶,得到中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III),產率86%。
(2)磷氯協同苯并吡喃類阻燃劑(化合物I)的制備
將4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)(0.10mol)和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物(DOPO,0.10mol)溶于100毫升甲苯中,攪拌回流反應4~7小時,反應完畢后,旋轉蒸發除去有機溶劑,析出白色固體,減壓抽濾,粗產品用甲苯-甲醇混合溶劑重結晶,真空干燥,得到目標產物,產率為75%。DSC測得熔點為226.57℃(峰值)。核磁共振氫譜圖和紅外光譜圖分別如圖1和圖2所示。
1H NMR(300MHz,DMSO-d6/TMS)δ:4.77-4.88(m,1H),4.99-5.09(m,2H),6.54-6.62(m,1H),6.97(d,J=8.4Hz,1H),7.23-7.46(m,5H),7.65-7.68(m,1H),7.86(t,J=7.8Hz,1H),7.94-8.01(m,1H),8.16-8.28(m,2H).IR(KBr)ν:3195,3073,2854,1633,1607,1594,1476,1431,1252,1229,1195,1119,1047,1020,924,825,758,553cm-1.
用Netzsch TG 209熱重分析儀測試了化合物的熱失重性能,見圖3。由圖3可見,該化合物在241℃失重為10%,380℃失重為50%,在600℃時殘炭率為20.79%,790℃時殘炭率仍高達15.64%,具有較好的熱穩定性和成炭性。
實施例2:磷氯協同苯并吡喃類阻燃劑的制備
(1)中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)
在干燥的三口燒瓶中加入DMF(0.15mol),冰水浴冷卻至0-5℃,在攪拌下緩慢滴加POCl3(0.15mol),滴加過程中控制反應溫度不超過5℃,滴加完畢后,維持0-5℃繼續攪拌反應1小時;然后,在10℃下,緩慢滴加6-氯-4-色滿酮(化合物II)的DMF溶液(0.10mol 6-氯-4-色滿酮溶于20毫升DMF),之后,在70-90℃反應3-5小時;反應完畢后,將反應液冷卻至室溫,在快速攪拌下,緩慢傾入冰水中,靜置,抽濾、水洗、干燥,經乙醇重結晶,得到中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III),產率78%。
(2)磷氯協同苯并吡喃類阻燃劑(化合物I)的制備
將4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)(0.10mmol)和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物(DOPO,0.08mmol)溶于100毫升甲苯中,攪拌回流反應4~7小時,反應完畢后,旋轉蒸發除去有機溶劑,析出白色固體,減壓抽濾,粗產品用甲苯-甲醇混合溶劑重結晶,真空干燥,得到目標產物,產率為73%。
實施例3:磷氯協同苯并吡喃類阻燃劑的制備
(1)中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)
在干燥的三口燒瓶中加入DMF(0.35mol),冰水浴冷卻至0-5℃,在攪拌下緩慢滴加POCl3(0.30mol),滴加過程中控制反應溫度不超過5℃,滴加完畢后,維持0-5℃繼續攪拌反應1小時;然后,在10℃下,緩慢滴加6-氯-4-色滿酮(化合物II)的DMF溶液(0.10mol 6-氯-4-色滿酮溶于20毫升DMF),之后,在70-90℃反應3-5小時;反應完畢后,將反應液冷卻至室溫,在快速攪拌下,緩慢傾入冰水中,靜置,抽濾、水洗、干燥,經乙醇重結晶,得到中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III),產率88%。
(2)磷氯協同苯并吡喃類阻燃劑(化合物I)的制備
將4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)(0.10mol)和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物(DOPO,0.12mol)溶于100毫升甲苯中,攪拌回流反應4~7小時,反應完畢后,旋轉蒸發除去有機溶劑,析出白色固體,減壓抽濾,粗產品用四氫呋喃-甲醇混合溶劑重結晶,真空干燥,得到目標產物,產率為70%。
實施例4:磷氯協同苯并吡喃類阻燃劑的制備
(1)中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)
在干燥的三口燒瓶中加入DMF(0.35mol),冰水浴冷卻至0-5℃,在攪拌下緩慢滴加POCl3(0.15mol),滴加過程中控制反應溫度不超過5℃,滴加完畢后,維持0-5℃繼續攪拌反應1小時;然后,在10℃下,緩慢滴加6-氯-4-色滿酮(化合物II)的DMF溶液(0.10mol 6-氯-4-色滿酮溶于20毫升DMF),之后,在70-90℃反應3-5小時;反應完畢后,將反應液冷卻至室溫,在快速攪拌下,緩慢傾入冰水中,靜置,抽濾、水洗、干燥,經乙醇重結晶,得到中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III),產率82%。
(2)磷氯協同苯并吡喃類阻燃劑(化合物I)的制備
將4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)(0.10mol)和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物(DOPO,0.10mmol)溶于100毫升二氧六環中,攪拌回流反應4~7小時,反應完畢后,旋轉蒸發除去有機溶劑,析出白色固體,減壓抽濾,粗產品用四氫呋喃-甲醇混合溶劑重結晶,真空干燥,得到目標產物,產率為73%。
實施例5:磷氯協同苯并吡喃類阻燃劑的制備
(1)中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)
在干燥的三口燒瓶中加入DMF(0.15mol),冰水浴冷卻至0-5℃,在攪拌下緩慢滴加POCl3(0.30mol),滴加過程中控制反應溫度不超過5℃,滴加完畢后,維持0-5℃繼續攪拌反應1小時;然后,在10℃下,緩慢滴加6-氯-4-色滿酮(化合物II)的DMF溶液(0.10mol 6-氯-4-色滿酮溶于20毫升DMF),之后,在70-90℃反應3-5小時;反應完畢后,將反應液冷卻至室溫,在快速攪拌下,緩慢傾入冰水中,靜置,抽濾、水洗、干燥,經乙醇重結晶,得到中間體4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III),產率78%。
(2)磷氯協同苯并吡喃類阻燃劑(化合物I)的制備
將4,6-二氯-2H-苯并吡喃-3-甲醛(化合物III)(0.10mol)和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物(DOPO,0.12mmol)溶于100毫升二氧六環中,攪拌回流反應4~7小時,反應完畢后,旋轉蒸發除去有機溶劑,析出白色固體,減壓抽濾,粗產品用甲苯-甲醇混合溶劑重結晶,真空干燥,得到目標產物,產率為71%。
實施例6:磷氯協同苯并吡喃類阻燃劑的阻燃性能測試
本發明將上述磷氯協同苯并吡喃類阻燃劑應用于環氧樹脂中,并采用極限氧指數法測定了該阻燃劑的阻燃性能。將環氧樹脂和阻燃劑以不同比例混合均勻,加熱至80℃后加入固化劑(固化劑與環氧樹脂的質量比為1:10)并攪拌均勻,然后注入模具中,固化2小時,之后再在120℃固化3小時,所得樣條參照GB/T 2406.2‐2009測定其極限氧指數,結果見表1所示。由表1可以看出,阻燃劑的添加能夠明顯提高環氧樹脂的阻燃性能,當阻燃劑添加量達到20wt%時,極限氧指數達到28%,具有良好的阻燃效果,屬難燃級別。因此,本發明的阻燃劑具有良好的阻燃性能。
表1阻燃性能測試結果
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種制備手性二氫1,4-苯并噁嗪類化合物的方法與流程
- 一種手性螺環羥吲哚?苯并吡喃?酮并?3,4?二氫?吡喃化合物的合成方法與流程
- 基于噻吩并異苯并吡喃不對稱給電子單元的D?A型聚合物光伏材料及其應用的制作方法與工藝
- 2?芳氧基羧酸(二氫苯并呋喃?7?氧基)烷基酯的制作方法與工藝
- 一種具有6H?二苯并吡喃結構化合物及其制備方法與流程
- 一種4H?苯并吡喃類化合物的制備方法與流程
- 一種6H?苯并[C]苯并吡喃及其衍生物的制備方法與流程
- 一種具有苯并吡喃結構的FSH拮抗劑的制備方法與流程
- 一種6H?苯并[C]苯并吡喃類化合物的合成方法與流程
- 一種3,4?二氫吡喃[3,2?c]色烯?5?酮類化合物的合成方法及其應用與流程