一種18F標記的多肽類腫瘤凋亡檢測試劑及其制備方法及應用與流程
本發明屬于放射性藥物及核醫學技術領域,具體涉及一種18f標記的多肽類腫瘤凋亡檢測試劑,并進一步公開其制備方法和應用。
背景技術:
誘導腫瘤細胞凋亡是多種腫瘤治療的主要作用機制之一,因此測定腫瘤細胞是否產生凋亡已成為評價腫瘤治療療效的一種重要指標。當前檢測腫瘤細胞凋亡的方法主要有體外、體內兩大類方法:傳統的體外檢測方法較為成熟,但只能檢測某一時間點的細胞凋亡狀態,不能實時、動態地檢測腫瘤細胞凋亡的發生和發展過程,因此限制了體外檢測方法的臨床廣泛應用。而體內檢測方法則由于可以動態監測細胞的凋亡情況,受到廣泛關注。
隨著分子影像技術的不斷發展,無創的放射性核素顯像已成為目前檢測腫瘤細胞凋亡的研究熱點,其中主要包括單光子發射計算機斷層成像(singlephotonemissioncomputedtomography,spect)與正電子發射斷層成像(positronemissiontomography,pet)。與spect顯像相比,pet顯像具有明顯的優勢,如靈敏度高、空間分辨率強,并能夠進行動態、定量、定位分析等優勢,因此,有關于腫瘤凋亡監測的pet顯像劑的研究越來越多。
目前,常用于pet顯像的凋亡顯像劑的主要靶點有磷酯酰絲氨酸(phosphatidylserine,簡稱ps)和半胱氨酸天冬氨酸蛋白酶(cysteinylaspartatespecificproteinase,簡稱caspase),但靶向ps的顯像劑難以區分腫瘤凋亡和壞死,識別靶點僅限于膜表面,因此顯像靈敏度相對不高;而caspase-3由于只存在凋亡細胞中,因此以caspase-3為靶點的pet顯像劑則比以ps為靶點的顯像劑的特異性更高。
caspase是一組存在于細胞質中具有類似結構的含半胱氨酸的天冬氨酸蛋白水解酶,參與細胞的生長、分化與凋亡調節,并且與細胞的凋亡密切相關。其中,caspase-3是起凋亡執行者的作用,在正常細胞中處于非活化的酶原狀態,而當細胞發生凋亡作用時則表現為激活狀態,而激活的caspase-3可以特異性地識別氨基酸序列結構asp-glu-val-asp(devd)。因此,以caspase-3為靶點,選擇可被其特異性識別的多肽序列devd為靶向基團制備新型腫瘤凋亡顯像探針得到了迅速發展,并且通過荷瘤鼠pet顯像也已經證實了它們在檢測細胞凋亡方面的優勢。
然而,這些標記探針在放射性標記方面則存在一些缺陷,首先在制備方法上,現有標記探針均是通過兩步法完成:首先利用親核取代反應從對甲苯磺酰基(ots)上將18f離子引入,然后通過“點擊化學”反應將標記基團與多肽序列相連,得到目標探針。其中,氟化過程需要在無水條件下進行,并需要半制備hplc法進行純化,反應條件較苛刻且步驟較為繁瑣,并導致標記時間過長。最近,zhiboliu等人成功地將一步18f-19f離子交換法運用到探針的制備中,簡化了探針的標記步驟,一定程度上縮短了合成時間,并且避免了半制備hplc的純化。同時,該標記方法所需的反應條件溫和,具有一定的通用性,已被運用到一系列放射性pet探針的制備中。
技術實現要素:
為此,本發明所要解決的技術問題在于提供一種18f標記的多肽化合物,并進一步公開其用于制備腫瘤凋亡檢測試劑的應用。
為解決上述技術問題,本發明所述的一種18f標記的多肽化合物,所述多肽化合物記為18f-ambf3-gdevd,具有如下所示的結構:
本發明還公開了所述18f標記的多肽化合物的標記前體,具有如下式(1)所示的結構:
本發明還公開了一種制備所述標記前體的方法,包括如下步驟:
(1)接氨基酸:取fmoc-l-天冬氨酸beta-叔丁酯溶解,在dipea存在下與樹脂進行反應;隨后抽干反應溶劑,并加入洗滌試劑進行洗滌,洗滌至無裸露的氨基;
(2)脫fmoc基團:向上述反應物中加入哌啶,抽干溶劑并洗滌至fmoc保護基脫除;
(3)取fmoc-l-纈氨酸加入上述反應物中,在hbtu和dipea存在下,重復上述步驟(1)-(2)進行氨基酸的連接以及fmoc基團的脫除;
(4)取fmoc-o-叔丁基-l-谷氨酸加入上述反應物中,在hbtu和dipea存在下,重復上述步驟(1)-(2)進行氨基酸的連接以及fmoc基團的脫除;
(5)取fmoc-l-天冬氨酸beta-叔丁酯加入上述反應物中,在hbtu和dipea存在下,重復上述步驟(1)-(2)進行氨基酸的連接以及fmoc基團的脫除;
(6)取n-boc-d-炔丙基甘氨酸加入上述反應物中,在hbtu和dipea存在下,重復上述步驟(1)進行氨基酸的連接,并加入含tfa的dcm溶液洗滌脫除樹脂;
(7)取上述反應物溶于有機溶劑,在tfa和tips存在下進行反應,脫除叔丁基;
(8)在氮氣保護下,向上述反應物中加入ambf3試劑,溶于dmf-h2o溶劑,在抗壞血酸鹽和銅催化劑存在下,于40-50℃下進行反應,并通過半制備型hplc進行分離純化,得到黃色固體粉末,即得式(1)所示標記前體;
所述ambf3試劑的結構如反應方程式所示,可以選用現有市售產品,或者可以根據現有文獻方法進行合成,產物為黃色油狀物,m=1.12g。
所述步驟(1)中,還包括對所述樹脂進行前處理的步驟,具體包括將選定樹脂溶于二氯甲烷溶劑的步驟,并抽干溶劑。
所述步驟(1)中,所述洗滌步驟具體包括:以體積比為17:2:1的dmf-ch3oh-dipea混合溶劑進行洗滌,抽干試劑后再加入dmf溶劑進行洗滌。
所述步驟(6)中,還包括提純反應物的步驟,具體包括:將反應物抽濾,取濾液旋干,加入乙醚溶劑洗滌,固液分離后取固體干燥,得到白色固體粉末。
所述步驟(7)中,還包括提純反應物的步驟,具體包括:取反應物旋蒸得到油狀液體,加入乙醚溶劑洗滌,固液分離后取固體干燥,得到灰色固體粉末。
所述步驟(7)中,所述有機溶劑包括dcm、thf或者ch3cn。
所述步驟(8)中,所述銅催化劑為cuso4·5h2o或六氟磷酸四(乙腈)銅(ⅰ)。
本發明還公開了一種制備所述18f標記的多肽化合物的方法,取所述標記前體化合物與18f離子,在加熱條件下,進行標記反應,制得所述的18f標記的多肽化合物18f-ambf3-gdevd。
本發明還公開了所述的18f標記的多肽化合物用于制備腫瘤凋亡檢測試劑的用途。
本發明還公開了一種腫瘤凋亡檢測試劑,包括所述的18f標記的多肽化合物。
本發明所述的18f標記的多肽化合物18f-ambf3-gdevd(探針18f-1),采用固相多肽合成的方法,以不溶樹脂作為固相載體,按順序連接fmoc-l-天冬氨酸beta-叔丁酯、fmoc-l-纈氨酸、fmoc-o-叔丁基-l-谷氨酸、fmoc-l-天冬氨酸beta-叔丁酯和n-boc-d-炔丙基甘氨酸,獲得gly-asp-glu-val-asp(gdevd),產物純度達98%以上;在合成所述標記前體化合物1時,“點擊化學”反應具有產率高,反應條件溫和,且產物易于分離的優點,故通過“點擊化學”反應使多肽2上的炔基與ambf3試劑上的疊氮在抗壞血酸鈉和五水硫酸銅的作用下發生環加成反應,成功引入了用于一步18f標記的基團,整個反應條件溫和,且所得產物產率較高、純度較高。
本文首先通過固相法成功合成了產率高、純度高的多肽,然后通過“點擊化學”引入18f標記基團,得到產率高的標記前體1,再進一步通過一步18f標記法成功制備了高產率、高純度的pet探針18f-1,整個反應簡化了18f-標記步驟,避免了標記過程中的半制備hplc純化,縮短了標記時間。
本發明所述的18f標記的探針具有良好的體外穩定性和親水性,同時含有可被caspase-3特異性識別的devd序列。用阿霉素對腫瘤細胞hela作用后可以看到明顯的凋亡特征,caspase-3水平隨藥物濃度增加而升高。所得探針18f-1在腫瘤細胞及腫瘤凋亡細胞中攝取有明顯的差異,不同作用時間點下相對攝取值達到凋亡前的2.5-3.0倍,說明探針能被caspase酶有效識別,且在較短的時間內能夠看到顯著的差異。這些特點均表明探針18f-1是一種潛在的靶向caspase-3的凋亡顯像探針,有較好的應用前景。
附圖說明
為了使本發明的內容更容易被清楚的理解,下面根據本發明的具體實施例并結合附圖,對本發明作進一步詳細的說明,其中,
圖1為標記前體化合物1的hplc分析圖;
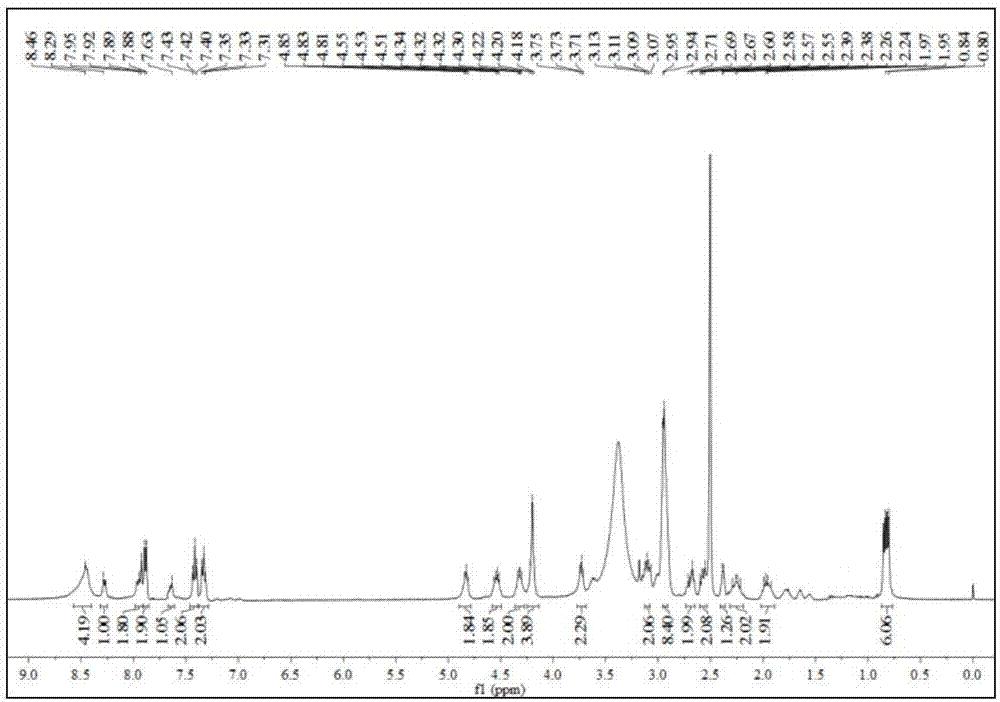
圖2為標記前體化合物1的核磁共振氫譜;
圖3為標記前體化合物1的核磁共振碳譜;
圖4為標記前體化合物1的核磁共振氟譜;
圖5為探針18f-1的放射性hplc分析圖;
圖6為標記產物18f-1在pbs與血清中孵育不同時間的放化純;
圖7為hela細胞在不同藥物濃度作用下的caspase3水平變化情況;
圖8為探針18f-1在腫瘤細胞及腫瘤凋亡細胞中攝取的差異情況。
具體實施方式
實施例1標記前體的合成
本實施例所述標記前體1的合成路線如下:
本實施例所述標記前體1的合成包括如下步驟:
(1)接氨基酸:稱取2-氯三苯甲基氯樹脂(544mg,loading值:1.106mmol/g)置于三通砂芯漏斗中,再加入10ml二氯甲烷(dcm)溶劑,振蕩10min,抽干溶劑;
稱取fmoc-l-天冬氨酸beta-叔丁酯(272mg,0.662mmol)溶于3mldmf溶劑,而后加入dipea(200μl,1.212mmol),超聲溶解后加入砂芯漏斗中與樹脂進行反應,并于室溫下振蕩2h;隨后抽干溶劑,并加入7ml溶液r(色譜純dmf:ch3oh:dipea=17:2:1)于三通砂芯漏斗中進行洗滌振蕩10min,抽干溶劑后,再用7mldmf溶劑洗滌,隨后抽干溶劑,并再加入上述溶液r進行洗滌,振蕩10min后抽干溶劑,并再加入7mldmf溶劑洗滌兩次,抽干溶劑;取一點樣品進行kaiser測試,顯示為淡黃色,說明沒有裸露的氨基;
(2)脫fmoc基團:向上述砂芯漏斗中加入7ml含20%哌啶的dmf溶液,振蕩10min后抽干溶劑,重復2次,再用7mldmf洗滌5次并抽干;直至進行kaiser測試,顯示為深藍色,說明有裸露的氨基,fmoc保護基已經脫除;
(3)稱取fmoc-l-纈氨酸(224mg,0.662mmol),hbtu(276mg,0.728mmol),dipea(200μl,1.212mmol),溶于dmf溶劑進行反應,按照上述步驟(1)-(2)中方法進行氨基酸的連接以及fmoc基團的脫除;
(4)稱取fmoc-o-叔丁基-l-谷氨酸(281mg,0.662mmol),hbtu(276mg,0.728mmol),dipea(200μl,1.212mmol),溶于dmf溶劑進行反應,按照上述步驟(1)-(2)中方法進行氨基酸的連接以及fmoc基團的脫除;
(5)稱取fmoc-l-天冬氨酸beta-叔丁酯(272mg,0.662mmol),hbtu(276mg,0.728mmol),dipea(200μl,1.212mmol),溶于dmf溶劑進行反應,按照上述步驟(1)-(2)中方法進行氨基酸的連接以及fmoc基團的脫除;
(6)稱取n-boc-d-炔丙基甘氨酸(244mg,0.728mmol),hbtu(276mg,0.728mmol),dipea(200μl,1.212mmol),溶于dmf溶劑進行反應,按照上述步驟(1)中方法進行氨基酸的連接,得到淡黃色固體;隨后向砂芯漏斗中加入含1wt%tfa的dcm溶液,經洗滌、抽濾,并收集濾液,直至樹脂出現深紅色且不褪色;隨后將濾液旋干,加入20ml乙醚溶液,經離心棄去上清液,收集沉淀,放入真空干燥箱干燥,得到白色固體粉末,檢測產物m=385mg,計算產率為60.5%;
(7)取上述得到的多肽反應物(200mg)于反應瓶中,加入2mldcm和2mltfa,進行反應20min后,加入50μltips,并繼續反應30min,以薄層色譜(tlc)法進行檢測(dcm:ch3oh=5:1)產物情況,至判定反應結束時停止反應,旋蒸得到油狀液體,加入10ml乙醚溶液洗滌,離心并棄去液體,將沉淀放入真空干燥箱干燥,得到灰色固體粉末產物,檢測m=156mg,計算產率為94.5%;
(8)在氮氣保護下,將上述所得反應物(116mg,0.146mmol),和ambf3試劑化合物4(43mg,0.219mmol),溶于3mldmf-h2o(1:1,v/v)溶劑中,將抗壞血酸鈉(58mg,0.292mmol)、cuso4·5h2o(73mg,0.292mmol)依次加入上述溶液中,于45℃下加熱反應30min;以tlc法檢測產物情況(dcm:ch3oh=5:1),至判定反應結束時停止反應;通過半制備型hplc(純化方法如下表1所示)進行分離純化,得到黃色固體粉末,檢測m=108mg,計算產率為74.6%。通過分析型hplc(方法如下表2所示)對終產物進行純度檢測,結果如附圖1所示,產物純度大于98%。
表1檢測前體1的半制備型hplc梯度淋洗方法
表2分析型hplc梯度淋洗方法
通過核磁、質譜和元素分析對制得的標記前體化合物1進行表征。以d6-dmso為溶劑,測定標記前體化合物1的核磁共振氫譜1hnmr、核磁共振碳譜13cnmr和核磁共振氟譜19fnmr,譜圖分別見圖2-4;以甲醇作為溶劑,采用質譜儀測定化合物的電噴霧質譜;用元素分析儀對標記前體化合物1進行元素分析。
以d6-dmso為溶劑測定標記前體1,核磁結果如下:1hnmr(d6-dmso,400mhz),δ(ppm):8.46(s,4h),8.29(s,1h),7.92(d,j=12.00hz,2h),7.88(d,j=4.00hz,2h),7.63(s,1h),7.42(t,j=6.00hz,2h),7.33(t,j=8.00hz,2h),4.83(m,2h),4.55(m,2h),4.32(m,2h),4.20(m,4h),3.73(t,j=8.00hz,2h),3.11(m,2h),2.95(m,8h),2.70(m,2h),2.58(m,2h),2.39(m,1h),2.26(m,2h),1.97(m,2h),0.84(m,j=4.00hz,6h);13cnmr(101mhz,d6-dmso),δ(ppm):174.56,172.68,172.15,172.03,171.60,171.19,170.97,170.25,156.31,144.26,141.13,128.10,127.57,126.82,125.81,124.19,120.55,66.28,63.91,57.55,54.87,53.98,53.47,52.46,50.20,47.01,46.16,41.79,36.36,31.32,30.61,28.54,27.73,22.66,19.55;19fnmr(376mhz,d6-dmso),δ(ppm):-135.8。
在1hnmr(圖4)中,δ7.63ppm為三氮唑環上氫的信號峰,這表明“點擊化學”反應成功;δ7.92,7.88,7.42,7.33ppm分別對應芴環上氫的信號峰,δ2.95ppm對應與季銨相連的兩個甲基上氫的信號峰,δ0.84ppm對應纈氨酸上與叔碳相連的兩個甲基上氫的信號峰。
13cnmr(圖5)中,δ124.19,128.10ppm對應三氮唑環上碳的信號峰,δ174.56,172.68,172.15,172.03,171.60,171.19,170.97,170.25,156.31ppm對應羰基上碳的信號峰,δ144.26,141.13,127.57,126.82,125.81,124.19ppm對應芴環上6組碳的信號峰。
19fnmr(圖6)中,δ-135.8ppm對應三氟硼酸鹽上氟的信號峰,δ-73.4ppm對應未除去的三氟乙酸中氟的信號峰。
核磁結果表明前體1的實際結構與預計的理論結構一致。
質譜分析結果esi-ms:測定值:970.52[m-f]+,1012.59[m+na]+;計算值:970.39[m-f]+,1012.38[m+na]+。
元素分析結果為:c43h55bf3n9o14:c52.74(52.18),h5.43(5.60),n12.33(12.74),括號中為理論值,分析結果表明實測值和理論值基本吻合。
可見,核磁、質譜和元素分析均表明所得化合物為目標化合物1。
實施例2多肽化合物18f-ambf3-gdevd的合成
通過電子轟擊富氧水h218o,得到18f離子,經陰離子交換柱(qma)吸附18f離子,用300μl噠嗪(ph=2.5)的緩沖液將18f離子從qma柱上洗脫,轉移到1ml的標記反應管中,加入10-20μl(25mmol/l,溶于dmf)的實施例1制得的標記前體化合物1,在80℃下,加熱30min反應,即得多肽化合物18f-ambf3-gdevd,記為探針18f-1。
取微量放射性溶液稀釋,通過放射性高效液相檢測前體的標記情況。向反應液中加入20ml去離子水稀釋,通過c18柱(seppakplusc-18)純化,用去離子水(10ml)洗滌三次,最后用0.5ml乙醇將標記產物淋洗到西林瓶中,加入4.5ml去離子水稀釋(乙醇含量≤10%)。取少量溶液稀釋,通過放射性高效液相檢測純化情況。
如圖5放射性hplc檢測顯示,標記產物的保留時間為16.8min,放化純(rcp)大于98%。標記產物的保留時間與標記前體在紫外吸收下的保留時間(tr=16.6min)基本一致,表明標記成功。經衰變校正后,標記產物的放射化學產率為40%,比活度為1.5ci/μmol。
在凋亡探針18f-1的制備過程中,18f標記總時間為40min。與用-ots制備凋亡探針的方法相比,通過一步18f標記法制備凋亡探針18f-1,簡化了標記步驟,標記過程中無需半制備hplc純化,大大縮短了標記時間,更適用于適合臨床應用。
實驗例
1、pbs和血清中穩定性測定
為了模擬體內環境,分別考察了18f-1在pbs(ph=7.4)與血清中的穩定性。取4份400μlpbs(ph=7.4)緩沖液與100μl標記產物18f-1溶液分別混合,在37℃下分別加熱0.5、1、2、3和4h。取少量溶液稀釋,通過放射性高效液相檢測標記產物的穩定性。
取4份400μl胎牛血清與100μl標記產物18f-1溶液分別混合,在37℃下分別孵育0.5、1、2、3和4h。到時間點后再加入500μl乙腈,用高速離心機在12000g/min下離心5min,取上層清液,用放射性高效液相檢測標記產物的穩定性。
通過放射性hplc檢測器檢測不同孵育時間點18f-1的放射性化學純度,結果如圖6所示,探針18f-1在4h內的放化純均大于96%。這表明該探針在體外具有良好的穩定性,有利于臨床使用,值得進一步的體內實驗研究。
2、脂水分配系數測定
脂水分配系數是考察藥物水溶性與脂溶性的重要參數。水溶性差的藥物在體內軟組織攝取較高,容易導致藥物顯像本底偏高,降低腫瘤顯像的效果。
將pbs緩沖液與等體積的正辛醇充分振蕩,使兩相相互飽和,在室溫下靜置24h,用分液漏斗將兩相分離。取1ml正辛醇、0.95mlpbs緩沖液和0.05ml標記產物18f-1溶液混合,用渦旋混合器振蕩5min,充分混勻。離心機4000g/min下離心5min,在有機相和水相中分別平行取3個樣品(100μl)于γ計數管中,用γ計數儀測定其放射性活度,每個樣品測定3次,計算脂水分配系數(logp值)。
通過放射性方法測定探針18f-1的脂水分配系數為logp=-0.995±0.103,這說明該探針具有良好的親水特性,預示著其他軟組織的攝取可能較低,組織靶向性可能較好。
3、細胞凋亡確定
試劑:ripa裂解液及active-caspase3抗體購自上海碧云天生物技術有限公司;β-actin抗體、hrp標記二抗、westernblottingluminol試劑為美國santacruz公司產品;蛋白酶和磷酸酶抑制劑購自美國thermofisher科技公司。
方法:將hela細胞以5×105個/孔的濃度鋪至6孔板,然后用不同濃度的adr作用24h,去除培養液,將細胞用冰浴pbs洗一遍,然后用含有蛋白酶和磷酸酶抑制劑的ripa裂解液于4℃裂解5分鐘。將上述裂解液于4℃條件下12000rpm離心10min,取上清,bca法測蛋白濃度,上樣40μg蛋白至12%sds-page進行電泳。電泳結束后,將蛋白轉移至pvdf膜,用tbs洗膜2次,每次10min,然后用tbs+0.1%tween-20+5%脫脂奶粉室溫封閉1h,封閉結束后,tbst洗膜10min,然后加入抗體稀釋液:tbst+5%脫脂奶粉+一抗,4℃孵育過夜。然后用tbst洗膜3次,每次10min,然后加入抗體稀釋液:tbst+5%脫脂奶粉+二抗,室溫孵育1h。然后用tbst洗膜3次,每次10min,使用westernblottingluminol試劑進行曝光,使用imagej軟件對條帶光密度進行分析。
測定hela細胞在不同藥物濃度作用下的caspase3水平變化情況,結果見圖7所示。可見,隨著濃度的增加,活化的天冬氨酸特異性半胱氨酸蛋白酶-3(active-caspase3)表達呈現顯著上升趨勢,表現出濃度依賴性。當所給藥物阿霉素的濃度達到1umol/l以上時,表達更為顯著。通過此測定,明確了細胞凋亡的給藥濃度應大于1umol/l。
4、細胞攝取實驗
取人的宮頸癌(hela)細胞以4×105/孔的密度接種到6孔板中,放入37℃培養箱中過夜培養。第二天將這些孔分成兩組,第一組細胞將原有培養基吸去換為新鮮培養基繼續孵育24h。第二組換為含2μm的阿霉素的培養基培養24h。而后將放射性藥物w43以1μci/孔的量加入到6孔板中,分別培養0.5、1、2、4h。在孵育結束后,吸去培養基,將細胞用pbs緩沖液沖洗3次。每孔加入1ml的胰酶消化液,收集細胞裂解液,用γ計數儀測量放射性計數。對每孔中的細胞進行蛋白濃度標準化后,得到正常細胞與凋亡細胞的攝取比例,測定結果見圖8所示。
通過細胞攝取實驗,對比探針18f-1在凋亡細胞和正常細胞中的攝取差異(如圖8),明顯可以看出,探針18f-1在凋亡細胞中的攝取高于其在正常細胞中的攝取,具有統計學意義,為后續的動物pet實驗提供了實驗依據。
顯然,上述實施例僅僅是為清楚地說明所作的舉例,而并非對實施方式的限定。對于所屬領域的普通技術人員來說,在上述說明的基礎上還可以做出其它不同形式的變化或變動。這里無需也無法對所有的實施方式予以窮舉。而由此所引伸出的顯而易見的變化或變動仍處于本發明創造的保護范圍之中。
- 還沒有人留言評論。精彩留言會獲得點贊!