一種腫瘤干細胞靶向脂質納米粒及制備方法與應用與流程
本發明屬靶向脂質納米粒及制備方法,主要涉及一種腫瘤干細胞靶向脂質納米粒及其制備方法。
技術背景
固體脂質納米粒(solidlipidnanoparticle,sln)是在20世紀90年代初發展起來的一種可替代乳劑、脂質體和聚合物納米粒的新型膠體給藥系統,屬于亞微粒給藥系統。sln易制備并可用于靜脈給藥,也可用于口服或局部給藥,適合多種給藥途徑。sln作為藥物載體可提高藥物的生物利用度,可控制藥物釋放以及發揮良好的靶向性。
核酸適配體(aptamer)是短的dna或rna片段,一般由12~30個堿基構成。適配體可形成特定結構的空間構型,與目標蛋白發生特異性結合。與抗體相比,適配體具備分子量小、低免疫原性、易獲得等優點。a15為rna適配體,可識別腫瘤干細胞標志物cd133分子,且有較強親和力和特異性。a15由15個核糖核苷酸組成,文獻報道,與單克隆抗體相比,該適配體在腫瘤細胞球里滲透能力強、保留時間長。
鹽霉素是一種聚醚類離子型抗生素,對大多數革蘭氏陽性菌和各種球蟲有比較強的抑制和殺滅作用。其臨床前及臨床研究數據較少,少數文獻報道其存在嚴重的肌肉和神經毒性,限制其主要應用于禽類疾病。2009年gupta等人通過高通量篩選法,發現鹽霉素具有殺傷乳腺癌干細胞的能力,降低乳腺癌干細胞比例的作用比常規化療藥紫杉醇高100倍。在其他腫瘤上,如慢性淋巴細胞白血病,也已被證明可特異性的殺傷腫瘤干細胞。
技術實現要素:
本發明的一個目的是提供一種腫瘤干細胞靶向的脂質納米粒,該納米粒由a15-聚乙二醇-十八醇嫁接物,單硬脂酸甘油酯,單硬脂酸聚乙二醇酯,鹽霉素組成,其中a15-聚乙二醇-十八醇嫁接物的重量百分比為9.1~17.9wt%、單硬脂酸甘油酯的重量百分比為69.5~72.0wt%,單硬脂酸聚乙二醇酯的重量百分比為3.6~7.6wt%,鹽霉素的重量百分比為3.6~7.6wt%。
本發明的第二個目的是提供所述腫瘤干細胞靶向脂質納米粒的制備方法,具體通過以下步驟實現:
(1)分別取20mg單硬脂酸甘油酯、1.0~2.0mg單硬脂酸聚乙二醇酯及1.0~2.0mg端氨基聚乙二醇-十八醇嫁接物,1.0~2.0mg鹽霉素置于2.0ml無水乙醇,水浴50℃加熱溶解,為有機相,超純水為分散相,磁力攪拌條件下,有機相注入分散相,繼續攪拌5min,得含游離氨基的脂質納米粒(nh2-peg-sln/sal);
(2)取游離氨基脂質納米粒(nh2-peg-sln/sal)分散液,焦碳酸二乙酯處理水稀釋納米粒濃度至100μg/ml。加入氨基摩爾1~2倍的n,n-二琥珀酰亞胺基碳酸酯(dsc),室溫攪拌(400rpm)反應9h。加入等摩爾數的氨基a15,繼續攪拌反應過夜。反應結束后,將反應液置于分子量為30kda的超濾離心管。離心,棄濾液,加入焦碳酸二乙酯處理水清洗兩次,除去未反應的適配體。未濾過液體即為a15-peg3400-sln/sal納米分散液。
本發明的第三個目的是提供端氨基聚乙二醇-十八醇嫁接物的合成方法,通過以下步驟實現:分別取含有端氨基端羧基的聚乙二醇(nh2-peg3400-cooh)、溶于無水二氯甲烷配成濃度10mg/ml的溶液,加入同摩爾量的十八醇(odc)。按照摩爾比十八醇:二環己基碳二亞胺:n-羥基琥珀酰亞胺1:1:1~1.5的比例加入二環己基碳二亞胺及n-羥基琥珀酰亞胺,室溫反應24h,反應結束后,將反應液滴加至8~10倍體積的冷無水乙醚,4℃過夜,使產物析出,離心收集沉淀,棄上清液,收集沉淀,40℃烘干,得端氨基聚乙二醇-十八醇嫁接物(nh2-peg3400-odc)。
本發明的第四個目的是提供所述的腫瘤干細胞靶向的脂質納米粒在制備給藥系統中的應用。所述腫瘤干細胞為cd133分子高表達的肝癌干細胞。所載藥物為腫瘤干細胞特異性藥物鹽霉素。
本發明選取的a15適配體,與肝癌腫瘤干細胞的表面分子標記物cd133分子,具有較強的親和力,可通過與cd133分子的特異性結合,靶向肝癌腫瘤干細胞。親水的含端氨基端羧基的聚乙二醇通過酯化反應鏈接十八醇,十八醇的長脂肪鏈段可插入脂質納米粒。該聚乙二醇的端氨基游離在脂質納米粒外周,與a15適配體的氨基通過n,n-二琥珀酰亞胺基碳酸酯化學連接,實現a15適配體修飾脂質納米粒。肝癌干細胞靶向脂質納米粒作為載體包封腫瘤干細胞特異性藥物鹽霉素,通過a15的靶向作用遞送至腫瘤干細胞,并改善鹽霉素的水難溶性及體內分布差的特點,增強鹽霉素藥效。
附圖說明
圖1是十八醇、端氨基端羧基聚乙二醇及端氨基聚乙二醇-十八醇嫁接物的紅外分析圖譜。
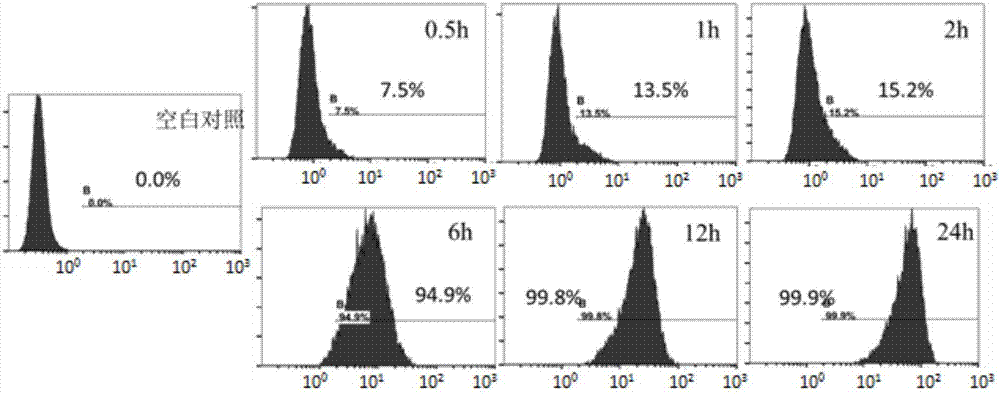
圖2是腫瘤干細胞球攝取該納米粒的細胞百分比及攝取熒光強度的流式分析圖,空白對照為未孵育納米粒組細胞。
具體實施方式
本發明結合附圖和實施例作進一步的說明。
一、端氨基聚乙二醇-十八醇嫁接物(nh2-peg3400-odc)制備
實施例1:
取200mgnh2-peg3400-cooh、15.6mg十八醇(odc),溶于20ml無水二氯甲烷,11.9mg加入二環己基碳二亞胺6.6mgn-羥基琥珀酰亞胺,室溫磁力攪拌(300rpm)反應24h。反應結束后,將反應液中滴加至的200ml冷無水乙醚。4℃過夜,使產物充分析出。4℃條件下3000rpm離心5min,收集沉淀,烘干,得端氨基聚乙二醇-十八醇嫁接物(nh2-peg3400-odc)。
實施例2:
取200mgnh2-peg3400-cooh、7.8mg十八醇(odc),溶于20ml無水二氯甲烷,加入11.9mg二環己基碳二亞胺10mgn-羥基琥珀酰亞胺,,室溫磁力攪拌(300rpm)反應24h。反應結束后,將反應液中滴加至的200ml冷無水乙醚。4℃過夜,使產物充分析出。4℃條件下3000rpm離心5min,收集沉淀,烘干,得端氨基聚乙二醇-十八醇嫁接物(nh2-peg3400-odc)。
實施例3:
取200mgnh2-peg3400-cooh、7.8mg十八醇(odc),溶于20ml無水二氯甲烷,加入11.9mg二環己基碳二亞胺7.9mgn-羥基琥珀酰亞胺,,室溫磁力攪拌(300rpm)反應24h。反應結束后,將反應液中滴加至的160ml冷無水乙醚。4℃過夜,使產物充分析出。4℃條件下3000rpm離心5min,收集沉淀,烘干,得端氨基聚乙二醇-十八醇嫁接物(nh2-peg3400-odc)。
傅里葉變換紅外光譜確證嫁接物結構,如附圖1。產物紅外光譜圖,720ppm位置處出現歸屬于十八醇的連續四個亞甲基(-(ch2)4-)的變形振動峰,且3350ppm處的羥基(-oh)伸縮振動峰消失,1650ppm吸收峰歸屬為新形成酯鍵(c=o)的伸縮振動峰。
二、游離氨基脂質納米粒(nh2-peg-sln/sal)制備
實施例1
取20mg單硬脂酸甘油酯、1.0mg鹽霉素(sal)、2mg單硬脂酸聚乙二醇酯及2mgnh2-peg3400-odc嫁接物,精密稱定,置于2.0ml無水乙醇,水浴50℃溶解,為有機相。20ml超純水為分散相,機械攪拌條件下(400rpm),有機相快速注入分散相,攪拌5min。飽和氯化鈉溶液0.5ml調節離子強度,絮凝納米粒,得含游離氨基脂質納米粒(nh2-peg-sln/sal)。經測定nh2-peg-sln/sal的粒徑及表面電位分別為
實施例2
取20mg單硬脂酸甘油酯、1.5mg鹽霉素(sal)、2.0mg單硬脂酸聚乙二醇酯及2mgnh2-peg3400-odc嫁接物,精密稱定,置于2.0ml無水乙醇,水浴50℃溶解,為有機相。20ml超純水為分散相,機械攪拌條件下(400rpm),有機相快速注入分散相,攪拌5min。飽和氯化鈉溶液0.5ml調節離子強度,絮凝納米粒,得含游離氨基脂質納米粒(nh2-peg-sln/sal)。經測定nh2-peg-sln/sal的粒徑90.2nm及表面電位為-9.5mv。
實施例3
取20mg單硬脂酸甘油酯、2.0mg鹽霉素(sal)、2.0mg單硬脂酸聚乙二醇酯及2.0mgnh2-peg3400-odc嫁接物,精密稱定,置于2.0ml無水乙醇,水浴50℃溶解,為有機相。20ml超純水為分散相,機械攪拌條件下(400rpm),有機相快速注入分散相,攪拌5min。飽和氯化鈉溶液0.5ml調節離子強度,絮凝納米粒,得含游離氨基脂質納米粒(nh2-peg-sln/sal)。經測定nh2-peg-sln/sal的粒徑93.2nm及表面電位為-10.2mv。
實施例4
取20mg單硬脂酸甘油酯、2.0mg鹽霉素(sal)、2.0mg單硬脂酸聚乙二醇酯及1.5mgnh2-peg3400-odc嫁接物,精密稱定,置于2.0ml無水乙醇,水浴50℃溶解,為有機相。20ml超純水為分散相,機械攪拌條件下(400rpm),有機相快速注入分散相,攪拌5min。飽和氯化鈉溶液0.5ml調節離子強度,絮凝納米粒,得含游離氨基脂質納米粒(nh2-peg-sln/sal)。經測定nh2-peg-sln/sal的粒徑為92.5nm及表面電位為-10.7mv。
實施例5
取20mg單硬脂酸甘油酯、2.0mg鹽霉素(sal)、2.0mg單硬脂酸聚乙二醇酯及1.0mgnh2-peg3400-odc嫁接物,精密稱定,置于2.0ml無水乙醇,水浴50℃溶解,為有機相。20ml超純水為分散相,機械攪拌條件下(400rpm),有機相快速注入分散相,攪拌5min。飽和氯化鈉溶液0.5ml調節離子強度,絮凝納米粒,得含游離氨基脂質納米粒(nh2-peg-sln/sal)。經測定nh2-peg-sln/sal的粒徑94.5nm及表面電位為-11.0mv。
實施例6
取20mg單硬脂酸甘油酯、2.0mg鹽霉素(sal)、1.5mg單硬脂酸聚乙二醇酯及2.0mgnh2-peg3400-odc嫁接物,精密稱定,置于2.0ml無水乙醇,水浴50℃溶解,為有機相。20ml超純水為分散相,機械攪拌條件下(400rpm),有機相快速注入分散相,攪拌5min。飽和氯化鈉溶液0.5ml調節離子強度,絮凝納米粒,得含游離氨基脂質納米粒(nh2-peg-sln/sal)。經測定nh2-peg-sln/sal的粒徑96.8nm及表面電位為-10.2mv。
實施例7
取20mg單硬脂酸甘油酯、2.0mg鹽霉素(sal)、1.0mg單硬脂酸聚乙二醇酯及2.0mgnh2-peg3400-odc嫁接物,精密稱定,置于2.0ml無水乙醇,水浴50℃溶解,為有機相。20ml超純水為分散相,機械攪拌條件下(400rpm),有機相快速注入分散相,攪拌5min。飽和氯化鈉溶液0.5ml調節離子強度,絮凝納米粒,得含游離氨基脂質納米粒(nh2-peg-sln/sal)。經測定nh2-peg-sln/sal的粒徑98.0nm及表面電位為-10.7mv。
實施例8
取20mg單硬脂酸甘油酯、1.0mg鹽霉素(sal)、1.0mg單硬脂酸聚乙二醇酯及2.0mgnh2-peg3400-odc嫁接物,精密稱定,置于2.0ml無水乙醇,水浴50℃溶解,為有機相。20ml超純水為分散相,機械攪拌條件下(400rpm),有機相快速注入分散相,攪拌5min。飽和氯化鈉溶液0.5ml調節離子強度,絮凝納米粒,得含游離氨基脂質納米粒(nh2-peg-sln/sal)。經測定nh2-peg-sln/sal的粒徑85.4nm及表面電位為-10.1mv。
三、肝癌干細胞靶向脂質納米粒制備
實施例1
取由20mg單硬脂酸甘油酯、2.0mg鹽霉素(sal)、2.0mg單硬脂酸聚乙二醇酯及2.0mgnh2-peg3400-odc嫁接物制備的游離氨基脂質納米粒(nh2-peg-sln/sal)分散液200μl。焦碳酸二乙酯處理水稀釋納米粒濃度至100μg/ml。加入14μl1.0mg/ml的n,n-二琥珀酰亞胺基碳酸酯(dsc)水溶液。室溫攪拌反應9h。加入54.9μl1mm的端氨基適配體a15-nh2,繼續攪拌反應過夜。反應結束后,將反應液置于分子量為30kda的超濾離心管。4℃條件下20000rpm離心10min,棄濾液,加入0.5ml焦碳酸二乙酯處理水清洗,重復該步驟兩次,除去未反應的適配體。未濾過液體即為a15-peg3400-sln/sal納米混懸液。經測定納米粒的粒徑為126nm,電位為-14.6mv。
實施例2
取由20mg單硬脂酸甘油酯、2.0mg鹽霉素(sal)、2.0mg單硬脂酸聚乙二醇酯及2.0mgnh2-peg3400-odc嫁接物制備的游離氨基脂質納米粒(nh2-peg-sln/sal)分散液200μl。焦碳酸二乙酯處理水稀釋納米粒濃度至100μg/ml。加入21μl1.0mg/ml的n,n-二琥珀酰亞胺基碳酸酯(dsc)水溶液。室溫攪拌反應9h。加入54.9μl1mm的端氨基適配體a15-nh2,繼續攪拌反應過夜。反應結束后,將反應液置于分子量為30kda的超濾離心管。4℃條件下20000rpm離心10min,棄濾液,加入0.5ml焦碳酸二乙酯處理水清洗,重復該步驟兩次,除去未反應的適配體。未濾過液體即為a15-peg3400-sln/sal納米混懸液。經測定納米粒的粒徑為106nm,電位為-16.2mv。
實施例3
取由20mg單硬脂酸甘油酯、2.0mg鹽霉素(sal)、2.0mg單硬脂酸聚乙二醇酯及2.0mgnh2-peg3400-odc嫁接物制備的游離氨基脂質納米粒(nh2-peg-sln/sal)分散液200μl。焦碳酸二乙酯處理水稀釋納米粒濃度至100μg/ml。加入28μl1.0mg/ml的n,n-二琥珀酰亞胺基碳酸酯(dsc)水溶液。室溫攪拌反應9h。加入54.9μl1mm的端氨基適配體a15-nh2,繼續攪拌反應過夜。反應結束后,將反應液置于分子量為30kda的超濾離心管。4℃條件下20000rpm離心10min,棄濾液,加入0.5ml焦碳酸二乙酯處理水清洗,重復該步驟兩次,除去未反應的適配體。未濾過液體即為a15-peg3400-sln/sal納米混懸液。經測定納米粒的粒徑為106nm,電位為-14.4mv.
連接a15適配體后,a15-peg3400-sln/sal的粒徑為略有增大;端氨基消失,納米粒電位下降。
四、腫瘤干細胞靶向脂質納米粒的腫瘤干細胞球攝取
實施例1:
取20mg單硬脂酸甘油酯、2mg單硬脂酸聚乙二醇酯,2mg鹽霉素及2mgnh2-peg3400-odc,精密稱定;另取100μg的硬脂胺-異硫氰酸熒光素,置于2.0ml無水乙醇,水浴50℃溶解,為有機相。20ml超純水為分散相,機械攪拌條件下(400rpm),有機相注入分散相,攪拌5min。飽和氯化鈉溶液調節分散液的離子強度,絮凝納米粒,制備熒光標記的游離氨基脂質納米粒
取制備的熒光標記nh2-peg-sln納米粒,焦碳酸二乙酯處理水稀釋納米粒濃度至100μg/ml。21μl1.0mg/ml的n,n-二琥珀酰亞胺基碳酸酯(dsc)溶液。室溫攪拌反應9h。加入54.9μl1mm的端氨基適配體a15-nh2,繼續攪拌反應過夜。反應結束后,將反應液置于分子量為30kda的超濾離心管。4℃條件下20000rpm離心10min,棄濾液,加入適量焦碳酸二乙酯處理水清洗,重復該步驟兩次,除去未反應的適配體。未濾過液體即熒光標記的a15-peg-sln/sal納米混懸液。
取免疫磁珠分選法分選得cd133+的肝癌腫瘤干細胞,并進行無血清懸浮球培養,待細胞球直徑生長至約200μm時,加入熒光標記的a15-peg-sln/sal納米混懸液,孵育不同時間(0.5h、2h、6h、12h)。
參見附圖2,流式細胞儀檢測攝取陽性細胞比例及細胞熒光強度,0.5h時7.5%的細胞可檢測到高于未孵育對照組細胞的熒光值,6h時94.9%的細胞有綠色熒光信號,幾乎細胞球所有細胞均有攝取。孵育時間繼續延長,熒光強度右移,細胞內攝取量不斷增多。a15修飾脂質納米粒形成的肝癌干細胞靶向脂質納米粒攝取遞送至腫瘤干細胞。
五、腫瘤干細胞靶向脂質納米粒作為藥物載體在制備抗腫瘤藥物中的應用
實施例1
采用酸性磷酸酶試驗法法,測定游離鹽霉素及肝癌干細胞靶向脂質納米粒a15-peg-sln/sal的腫瘤干細胞抗增殖活性。取分選得cd133+腫瘤干細胞,無血清懸浮球培養,待細胞球直徑生長至約200μm時,加入a15-peg-sln/sal納米粒及鹽霉素二甲亞砜溶液。37℃條件下與細胞球孵育48h。測定405nm處的吸光度值并計算細胞存活率。
肝癌腫瘤干細胞靶向脂質米粒的腫瘤干細胞ic50值為0.69±0.015μg/ml。鹽霉素組細胞存活率隨濃度的增加而下降,濃度為0.8μg/ml時,存活率降至55%時,繼續增加鹽霉素濃度,因受制于鹽霉素溶解性,藥物析出,細胞存活率改變不明顯。研究結果表明,肝癌腫瘤干細胞靶向脂質納米粒,可改善鹽霉素的溶解性,作為藥物載體可增強鹽霉素藥效。
- 還沒有人留言評論。精彩留言會獲得點贊!