一種抗菌疏水性生物可降解膜的制備方法與流程
技術領域
本發明涉及復合膜技術領域,具體涉及一種抗菌疏水性生物可降解膜的制備方法。
背景技術:
長期以來,以石油為原料的薄膜,在食品包裝以及其他工業領域占著主導地位,例如生物醫藥、廢水處理和化妝品等。盡管他們具有良好的機械性能和韌性,制備過程簡單且廉價,但是合成的這種聚合物難以降解,對環境造成嚴重污染,因此更多研究者已經利用可降解性聚合物制備。
殼聚糖是一種天然可降解生物材料,在農業、食品、化妝品、印染、醫學等領域均有廣泛應用。殼聚糖是甲殼素脫乙酰基的衍生物,而甲殼素是甲殼類動物及昆蟲細胞壁的主要成分,產量僅次于纖維素,是地球上第二大可再生資源。殼聚糖可以制備溶液、凝膠、海綿狀以及薄膜等,但其存在著缺點是溶液和凝膠易流動,不易在局部形成較高濃度,薄膜以及海綿狀物其機械性能和韌度較差。已有文獻報道,用戊二醛、乙二醛、雙官能團酸酐等交聯劑與殼聚糖交聯,可提高膜的機械強度,但醛類交聯劑與殼聚糖生成的Schiff堿具有化學不穩定性;雙官能團酸酐與殼聚糖交聯需在高溫下進行,易使殼聚糖降解;而且,殼聚糖分子內大量的羥基和氨基形成了復雜的分子內和分子間氫鍵,使其不溶于水和堿性溶液,只能溶于弱酸性溶液,這限制了它在更廣泛pH范圍的應用。
羧甲基纖維素鈉因具有良好的成膜性、生物降解性和生物相容性被廣泛應用于食品涂膜保鮮中。但單一的羧甲基纖維素鈉膜質脆、耐水性差、抑菌效果不明顯,限制了其在保鮮方面的應用。雖然已有將海藻酸鈉與羧甲基纖維素鈉共混制作食品包裝膜的報道,但是制得的海藻酸鈉與羧甲基纖維素鈉共混膜,力學性能較差,只能做一些食品的內包裝。
技術實現要素:
針對上述現有技術的缺陷,本發明的其中一個目的是提供一種抗菌疏水性生物可降解膜的制備方法,包括下列步驟:
1)稱取一定量的改性殼聚糖粉末和羧甲基纖維素鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫靜置溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為0.5~5%的改性殼聚糖溶液和質量分數為0.5~5%的羧甲基纖維素鈉溶液;
2)將上述配置的改性殼聚糖溶液與羧甲基纖維素鈉溶液按一定溶質質量配比在一定條件下混合,制得均一的改性殼聚糖/羧甲基纖維素鈉水溶液,然后將混合溶液倒入膜具,放入鼓風干燥箱干燥,待溶劑完全蒸發后,即得到干的改性殼聚糖/羧甲基纖維素鈉復合膜。
優選的,在步驟2)中,所述的改性殼聚糖溶液與羧甲基纖維素鈉溶液的溶質質量比為0.5:10~3:10,更為優選的為1:10~2:10,例如1:10~1.2:10、1:10~1.5:10、1:10~1.8:10、1.2:10~1.5:10、1.2:10~1.8:10、1.2:10~2:10、1.5:10~1.8:10、1.5:10~2:10或1.8:10~2:10。
優選的,在步驟2)中,所述的改性殼聚糖溶液與羧甲基纖維素鈉溶液混合時的溫度為15~60 ℃。
優選的,在步驟2)中,所述的改性殼聚糖溶液與羧甲基纖維素鈉溶液混合時的溫度為20~50 ℃,更為優選的為25~40 ℃,例如25~30 ℃、25~35 ℃、30~35 ℃、30~40 ℃、35~40 ℃、25 ℃、26 ℃、27 ℃、28 ℃、29 ℃、30 ℃、31 ℃、32 ℃、33 ℃、34 ℃、35 ℃、36 ℃、37 ℃、38 ℃、39 ℃或40 ℃。
優選的,在步驟2)中,所述干燥條件為干燥溫度35~60 ℃,干燥時間24~120 h。
優選的,在步驟2)中,所述干燥條件為干燥溫度35~55 ℃,更為優選的為35~45 ℃,例如35~40 ℃、40~45 ℃、35 ℃、36 ℃、37 ℃、38 ℃、39 ℃、40 ℃、41 ℃、42 ℃、43 ℃、44 ℃或45 ℃;干燥時間36~90 h,更為優選的為48~90 h,例如48~60 h、48~72 h、48~90 h、60~72 h、60~90 h、72~90 h、48 h、50 h、55 h、60 h、65 h、70 h、72 h、75 h、80 h、85 h或90 h。
優選的,在步驟1)中,所述改性殼聚糖粉末的取代度為0.56~0.93,更為優選的為0.70~0.93,例如0.70~0.75、0.70~0.80、0.70~0.85、0.70~0.90、0.75~0.80、0.75~0.85、0.75~0.90、0.75~0.93、0.80~0.85、0.80~0.90、0.80~0.93、0.85~0.90、0.85~0.93、0.70、0.71、0.72、0.73、0.74、0.75、0.76、0.77、0.78、0.79、0.80、0.81、0.82、0.83、0.84、0.85、0.86、0.87、0.88、0.89、0.90、0.91、0.92或0.93。
優選的,在步驟1)中,所述改性殼聚糖粉末由烷基季銨鹽與殼聚糖反應制得,或由烷基季銨鹽與氧取代殼聚糖羧酸鹽反應制得。
優選的,所述氧取代殼聚糖羧酸鹽為O-烷基羧基殼聚糖,其中所述烷基為甲基、乙基或丙基。
優選的,所述烷基季銨鹽為2,3-環氧丙基三甲基氯化銨、2,3-環氧丙基二甲基癸基氯化銨、2,3-環氧丙基二甲基十二烷基氯化銨、2,3-環氧丙基二甲基十四烷基氯化銨、2,3-環氧丙基二甲基十六烷基氯化銨、2,3-環氧丙基二甲基十八烷基氯化銨或3-氯-2-羥丙基三甲基氯化銨中的一種。
優選的,所述改性殼聚糖粉末通過以下一種方法制得:
a)將殼聚糖加入到離子液體中,于60~100 ℃,0.01~0.06 MPa負壓的條件下,恒溫攪拌1~6 h,使殼聚糖溶解于離子液體中,所述殼聚糖在殼聚糖/離子液體溶液中的質量百分比濃度為0.5~2.5%;
b)向所述殼聚糖/離子液體溶液中加入所述烷基季銨鹽,于60~120 ℃,0.01~0.06 MPa負壓的條件下,反應0.5~10 h,所述n(烷基季銨鹽):n(殼聚糖)為1:1~6:1,其中n(殼聚糖)指的是殼聚糖的重復單元數;
c)反應結束后,向冷卻至室溫的步驟2)的反應液中加入丙酮-乙醇混合溶液,離心析出沉淀物,沉淀物用所述丙酮-乙醇混合溶液重復洗滌2~3次,并于50~80 ℃條件下干燥8~24 h,得到氮取代殼聚糖季銨鹽;
d)將混有丙酮和乙醇的離子液體,蒸餾分離,分別回收丙酮、乙醇和離子液體;
其中所述離子液體為1-丁基-3-甲基氯化咪唑、1-丁基-3-甲基溴化咪唑、1-烯丙基-3-甲基氯化咪唑或1-乙基-3-甲基咪唑醋酸鹽中的一種。
優選的,在步驟b)中,所述的n(烷基季銨鹽):n(殼聚糖)為2:1~4:1,反應溫度為80~100 ℃,反應時間為6~8 h,所述n(殼聚糖)指的是殼聚糖的重復單元數。
優選的,在步驟a)中,所述的溶解溫度為80 ℃,溶解時間4 h。
優選的,所述殼聚糖的脫乙酰度為75~95%,平均分子量為200~2000 mPa.s。
優選的,所述步驟c)的丙酮-乙醇混合溶液中,丙酮與乙醇的體積比為1:1~1:4。
優選的,在步驟c)中,所述的干燥溫度為60 ℃,干燥時間為12 h。
優選的,所述步驟d)具體操作為:將混有乙醇和丙酮的離子液體溶液置于電加熱套上,常壓蒸餾冷凝分離丙酮和乙醇;回收離子液體,用乙酸乙酯萃取后,三次旋蒸除去多余的乙酸乙酯,于60~100 ℃的真空干燥箱中0.06~0.1 MPa負壓下干燥10~24 h。
一種通過另一種方法制得的改性殼聚糖粉末,在上述改性殼聚糖粉末制備方法中,用氧取代殼聚糖羧酸鹽替換所述殼聚糖。
本發明的另一目的是提供一種抗菌疏水性生物可降解膜的制備方法,用海藻酸鈉替換上述制備方法中的羧甲基纖維素鈉。
本發明中所述羧甲基纖維素鈉為USP級,粘度400~1400 mPa.s;所述海藻酸鈉購于青島明月海藻集團有限公司,粘度分為60 mPa.s、370 mPa.s、540 mPa.s三種;其余所用原料、試劑等未注明生產廠商者,均為可以通過市購獲得的常規產品。
本發明另一目的在于提供一種利用上述方法制備得到的抗菌疏水性生物可降解膜。
本發明的又一目的在于提供上述抗菌疏水性生物可降解膜在食品包裝,特別是在濕食品的包裝中的應用。
本發明制備的抗菌疏水性生物可降解膜的力學性能測試:采用濟南天辰公司的電子萬能拉力機測試本發明制備的抗菌疏水性生物可降解膜的拉伸長度以及斷裂伸長率。具體操作為:選擇均勻、干凈、無瑕疵的本發明制備的樣品及對比樣品純的HTCC膜、CMC膜。樣品的厚度約0.07 mm,將樣品裁剪成長方形,長100 mm,寬16 mm。測試時夾距50 mm,拉伸速率10 mm/min。
本發明制備的抗菌疏水性生物可降解膜的抗菌性能測試:大腸桿菌和金黃色葡萄球菌是常見的革蘭氏陰性菌和革蘭氏陽性菌,因此選這2種菌來測試本發明制備的抗菌疏水性生物可降解膜的抗菌性能。具體操作為將定量濾紙片裁剪成8mm大小的圓片,放入滅菌鍋里121 ℃下滅菌20 min。在潔凈工作臺上將滅菌過的濾紙片,沾取不同溶質質量配比的復合膜溶液樣品及對比樣品溶液,并用玻片壓實,然后小心的貼在平板培養基表面上,最后將平板置于生化培養箱中于36 ℃培養24 h,測試樣品的抗菌性能。
與現有技術相比,本發明提供的抗菌疏水性生物可降解膜的制備方法利用的是改性殼聚糖與羧甲基纖維素鈉或海藻酸鈉反應,而非傳統的殼聚糖,由于改性的殼聚糖中接入季銨鹽基團,賦予殼聚糖良好的水溶性,這使得該方法在操作過程中無需通過加鹽酸、醋酸等方法使殼聚糖質子化、溶解,簡便了反應操作,消除了對反應設備的酸性腐蝕損耗;而且由本發明提供的方法制備得到的復合膜具有良好的生物可降解性、殺菌抑菌性能及疏水性,與不具備殺菌抑菌性、疏水性的羧甲基殼聚糖膜形成鮮明的對比;同時本發明制備的復合膜的抗張強度和斷裂伸長率與單一的羧甲基纖維素膜、海藻酸鈉膜或改性殼聚糖膜相比均有顯著增加。
附圖說明
圖1為本發明實施例2~4制備的2,3-環氧丙基三甲基氯化銨改性殼聚糖/羧甲基纖維素鈉膜(HTCC/CMC膜)及純的HTCC膜、純的CMC膜的透光性對比圖,其中a)代表純CMC膜的透光性對比圖;b)代表HTCC與CMC質量比為0.05的膜的透光性對比圖;c)代表HTCC與CMC質量比為0.10的膜的透光性對比圖;d)代表HTCC與CMC質量比為0.15的膜的透光性對比圖;e)代表純HTCC膜的透光性對比圖。
圖2為本發明實施例2~4制備的HTCC/CMC膜及純的HTCC膜、純CMC膜的切面的掃描電子顯微鏡照片,其中a)代表純CMC膜的電鏡照片;b)代表HTCC與CMC質量比為0.05的膜的電鏡照片;c)代表HTCC與CMC質量比為0.10的膜的電鏡照片;d)代表HTCC與CMC質量比為0.15的膜的電鏡照片;e)代表純HTCC膜的電鏡照片。
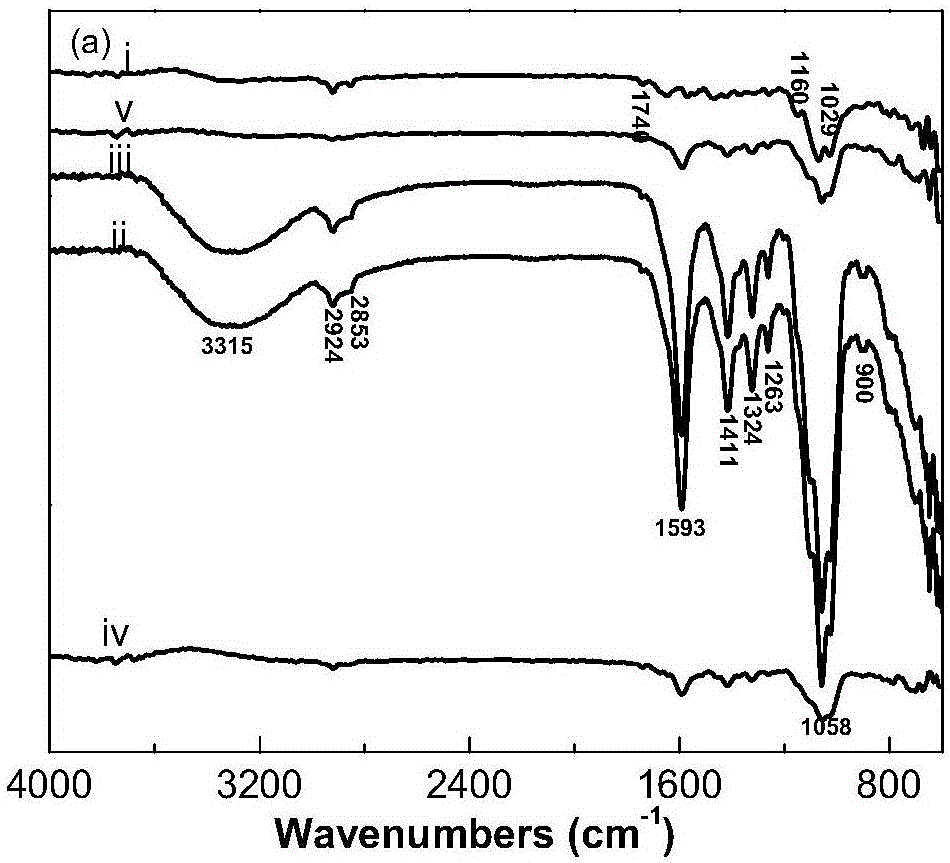
圖3為本發明實施例2~4制備的HTCC/CMC膜的紅外譜圖,其中i)代表純CMC膜的紅外譜圖;ii)代表HTCC與CMC質量比為0.05的膜的紅外譜圖;iii)代表HTCC與CMC質量比為0.10的膜的紅外譜圖;iv)代表HTCC與CMC質量比為0.15的膜的紅外譜圖;v)代表純HTCC膜的紅外譜圖。
圖4為本發明實施例2~4制備的HTCC/CMC膜及純的HTCC膜、純CMC膜的應力-應變曲線,其中i)代表純CMC膜的應力-應變曲線;ii)代表HTCC與CMC質量比為0.05的膜的應力-應變曲線;iii)代表HTCC與CMC質量比為0.10的膜的應力-應變曲線;iv)代表HTCC與CMC質量比為0.15的膜的應力-應變曲線;v)代表純HTCC膜的應力-應變曲線。
圖5為本發明實施例2~4制備的HTCC/CMC膜及純的HTCC膜、純的CMC膜的接觸角圖,其中a)代表純HTCC膜的接觸角圖;b)代表HTCC與CMC質量比為0.05的膜的接觸角圖;c)代表HTCC與CMC質量比為0.10的膜的接觸角圖;d)代表HTCC與CMC質量比為0.15的膜的接觸角圖;e)代表純CMC膜的接觸角圖。
圖6為本發明實施例2~6制備的HTCC/CMC膜及純的HTCC膜對金黃色葡萄球菌(黑色方形點)和大腸桿菌(紅色圓形點)的抗菌活性結果圖。
具體實施方式
下面結合具體實施例,對本發明進行詳細描述。實施例中未注明具體條件者,按照常規條件或制造商建議的條件進行。
實施例1
下面實施例用到的改性殼聚糖按以下步驟制備得到:
在100 mL的單口燒瓶中加入20 g離子液體1-丁基-3-甲基溴化咪唑和0.5 g黏度為300 mPa.s的殼聚糖,所述殼聚糖為經氫溴酸預處理并冷凍干燥制得,將單口燒瓶抽真空后密封,在0.06 MPa負壓條件下,攪拌均勻,置于100 ℃的油浴中恒溫攪拌4 h。通過偏光顯微鏡觀察殼聚糖完全溶解后,向單口燒瓶中加入n(烷基季銨鹽) : n(殼聚糖)為6:1的環氧當量為0.85的2,3-環氧丙基三甲基氯化銨(GTMAS),重新保持0.06 MPa負壓并將單口燒瓶置于80 ℃的油浴中恒溫反應8 h,以保證殼聚糖和GTMAS充分反應。
反應完成,向單口燒瓶中加入60 mL的丙酮-乙醇混合液,所述混合液中丙酮與乙醇的體積比為1:4,充分攪拌、靜置,使殼聚糖季銨鹽析出,以同樣的方法重復洗滌產物2次。最終產物置于60 ℃的真空干燥箱中干燥24 h,即得環氧丙基三甲基氯化銨改性殼聚糖。
洗滌液回收、蒸餾,分兩段溫度分別收集55-56 ℃的餾分丙酮和78-79 ℃的餾分乙醇,殘液為離子液體。將殘液離子液體用乙酸乙酯萃取后,三次旋蒸除去多余的乙酸乙酯,于60 ℃的真空干燥箱中,0.06 MPa負壓下干燥24 h。
本實施例中制得的改性殼聚糖的取代度為0.93。
實施例2
稱取一定量的取代度為0.56的HTCC粉末和CMC粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為2%的HTCC溶液和質量分數為2%的CMC溶液;
按溶質質量比為0.5:10的比例稱取上述配置的HTCC溶液與CMC溶液,并于25 ℃水浴下混合,制得均一的HTCC/CMC水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱45 ℃干燥24 h,待溶劑完全蒸發后,即得到干的HTCC/CMC復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的HTCC分子和CMC分子之間存在良好的相容性和顯著的相互作用。復合膜的抗張強度為26.45 MPa,與純CMC膜的11.95 MPa相比,其抗張強度增加了121%;斷裂伸長率由純CMC膜的1.55%增加至2.82%。室溫下,水在HTCC/CMC復合膜的接觸角為119o,而水在純CMC膜的接觸角僅為72.5o。根據文獻報道,單一的CMC膜沒有殺菌抑菌性,純HTCC膜對金黃色葡萄球菌和大腸桿菌的抑制圈直徑分別為1.23 cm和1.07 cm,而HTCC/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為1.13 cm,對大腸桿菌的抑菌圈直徑為1.00 cm,與純HTCC膜對上述兩種菌的抑制能力相當。
實施例3
實施條件與步驟同實施案例2,只是將配置好的HTCC溶液與CMC溶液的溶質質量比0.5:10替換為1:10。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的HTCC分子和CMC分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為28.56 MPa,比純CMC膜的抗張強度增加了139%;斷裂伸長率為2.86%;水在復合膜的接觸角為90o,大于在純CMC膜的接觸角,表現出良好的疏水性;HTCC/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為1.28 cm,對大腸桿菌的抑菌圈直徑為1.03 cm。
實施例4
實施條件與步驟同實施案例2,只是將配置好的HTCC溶液與CMC溶液的溶質質量比0.5:10替換為1.5:10。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的HTCC分子和CMC分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為21.82 MPa,比純CMC膜的抗張強度增加了82.6%;斷裂伸長率為2.27%;水在復合膜的接觸角為117o,大于在純CMC膜的接觸角,表現出良好的疏水性;HTCC/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為1.43 cm,對大腸桿菌的抑菌圈直徑為1.07 cm。
實施例5
實施條件與步驟同實施案例2,只是將配置好的HTCC溶液與CMC溶液的溶質質量比0.5:10替換為2:10。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的HTCC分子和CMC分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為27.82 MPa,比純CMC膜的抗張強度增加了132.8%;斷裂伸長率為2.57%;水在復合膜的接觸角為114o,大于在純CMC膜的接觸角,表現出良好的疏水性;HTCC/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為1.53 cm,對大腸桿菌的抑菌圈直徑為1.10 cm。
實施例6
實施條件與步驟同實施案例2,只是將配置好的HTCC溶液與CMC溶液的溶質質量比0.5:10替換為2.5:10。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的HTCC分子和CMC分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為28.95 MPa,比純CMC膜的抗張強度增加了142.2%;斷裂伸長率為3.02%;水在復合膜的接觸角為120o,大于在純CMC膜的接觸角,表現出良好的疏水性;HTCC/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為1.67 cm,對大腸桿菌的抑菌圈直徑為1.17 cm。
實施例7
稱取一定量的取代度為0.56的2-羥基丙基-N-二甲基十八烷基氯化銨改性殼聚糖粉末和海藻酸鈉(Sodium alginate, SA)粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為0.5的2-羥基丙基-N-二甲基十八烷基氯化銨改性殼聚糖溶液和質量分數為0.5的海藻酸鈉溶液;
按溶質質量比為0.5:10的比例稱取上述配置的2-羥基丙基-N-二甲基十八烷基氯化銨改性殼聚糖溶液與海藻酸鈉溶液,并于25 ℃水浴下混合,制得均一的2-羥基丙基-N-二甲基十八烷基氯化銨改性殼聚糖/SA水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱55 ℃干燥90 h,待溶劑完全蒸發后,即得到干的2-羥基丙基-N-二甲基十八烷基氯化銨改性殼聚糖/SA復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的2-羥基丙基-N-二甲基十八烷基氯化銨改性殼聚糖分子和海藻酸鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為17.32 MPa,比純SA膜的抗張強度增加了102.3%;斷裂伸長率為5.81%。水在復合膜的接觸角為93o,大于在純SA膜的接觸角,表現出良好的疏水性。2-羥基丙基-N-二甲基十八烷基氯化銨改性殼聚糖/SA復合膜對金黃色葡萄球菌的抑菌圈直徑為1.17 cm,對大腸桿菌的抑菌圈直徑為1.15厘米。
實施例8
稱取一定量的取代度為0.65的2-羥基丙基-N-二甲基十二烷基氯化銨改性殼聚糖粉末和羧甲基纖維素鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為1.5的2-羥基丙基-N-二甲基十二烷基氯化銨改性殼聚糖溶液和質量分數為1.5的羧甲基纖維素鈉溶液;
按溶質質量比為1.5:10的比例稱取上述配置的2-羥基丙基-N-二甲基十二烷基氯化銨改性殼聚糖溶液與羧甲基纖維素鈉溶液,并于15 ℃水浴下混合,制得均一的2-羥基丙基-N-二甲基十二烷基氯化銨改性殼聚糖/CMC水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱37 ℃干燥36 h,待溶劑完全蒸發后,即得到干的2-羥基丙基-N-二甲基十二烷基氯化銨改性殼聚糖/CMC復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的2-羥基丙基-N-二甲基十二烷基氯化銨改性殼聚糖分子和羧甲基纖維素鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為31.53MPa,比純CMC膜的抗張強度增加了163.8%;斷裂伸長率為10.81%。水在復合膜的接觸角為133o,大于在純CMC膜的接觸角,表現出良好的疏水性。2-羥基丙基-N-二甲基十二烷基氯化銨改性殼聚糖/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為2.07 cm,對大腸桿菌的抑菌圈直徑為1.63 cm。
實施例9
稱取一定量的取代度為0.80的2-羥基丙基-N-二甲基癸基氯化銨改性殼聚糖粉末和海藻酸鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為2.5的2-羥基丙基-N-二甲基癸基氯化銨改性殼聚糖溶液和質量分數為2.5的海藻酸鈉溶液;
按溶質質量比為2:10的比例稱取上述配置的2-羥基丙基-N-二甲基癸基氯化銨改性殼聚糖溶液與海藻酸鈉溶液,并于50 ℃水浴下混合,制得均一的2-羥基丙基-N-二甲基癸基氯化銨改性殼聚糖/SA水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱48 ℃干燥100 h,待溶劑完全蒸發后,即得到干的2-羥基丙基-N-二甲基癸基氯化銨改性殼聚糖/SA復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的2-羥基丙基-N-二甲基癸基氯化銨改性殼聚糖分子和海藻酸鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為19.20MPa,比純SA膜的抗張強度增加了124.3%;斷裂伸長率為9.81%。水在復合膜的接觸角為123o,大于在純SA膜的接觸角,表現出良好的疏水性。2-羥基丙基-N-二甲基十八烷基氯化銨改性殼聚糖/SA復合膜對金黃色葡萄球菌的抑菌圈直徑為2.17 cm,對大腸桿菌的抑菌圈直徑為1.65 cm。
實施例10
稱取一定量的取代度為0.70的3-氯-2-羥丙基三甲基氯化銨改性殼聚糖粉末和羧甲基纖維素鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為3.5的3-氯-2-羥丙基三甲基氯化銨改性殼聚糖溶液和質量分數為3.5的羧甲基纖維素鈉溶液;
按溶質質量比為1.2:10的比例稱取上述配置的3-氯-2-羥丙基三甲基氯化銨改性殼聚糖溶液與羧甲基纖維素鈉溶液,并于60 ℃水浴下混合,制得均一的3-氯-2-羥丙基三甲基氯化銨改性殼聚糖/CMC水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱38 ℃干燥48 h,待溶劑完全蒸發后,即得到干的3-氯-2-羥丙基三甲基氯化銨改性殼聚糖/CMC復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的3-氯-2-羥丙基三甲基氯化銨改性殼聚糖分子和CMC分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為28.05 MPa,比純CMC膜的抗張強度增加了134.7%;斷裂伸長率為3.82%。水在復合膜的接觸角為118o,大于在純CMC膜的接觸角,表現出良好的疏水性。3-氯-2-羥丙基三甲基氯化銨改性殼聚糖/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為1.79 cm,對大腸桿菌的抑菌圈直徑為1.28 cm。
實施例11
稱取一定量的取代度為0.93的2-羥基丙基-N-二甲基十四烷基氯化銨改性殼聚糖粉末和海藻酸鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為4.5的2-羥基丙基-N-二甲基十四烷基氯化銨改性殼聚糖溶液和質量分數為4.5的海藻酸鈉溶液;
按溶質質量比為1:10的比例稱取上述配置的2-羥基丙基-N-二甲基十四烷基氯化銨改性殼聚糖溶液與海藻酸鈉溶液,并于35 ℃水浴下混合,制得均一的2-羥基丙基-N-二甲基十四烷基氯化銨改性殼聚糖/SA水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱52 ℃干燥80 h,待溶劑完全蒸發后,即得到干的2-羥基丙基-N-二甲基十四烷基氯化銨改性殼聚糖/SA復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的2-羥基丙基-N-二甲基十四烷基氯化銨改性殼聚糖分子和海藻酸鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為23.20 MPa,比純SA膜的抗張強度增加了171.0%;斷裂伸長率為9.91%。水在復合膜的接觸角為130o,大于在純SA膜的接觸角,表現出良好的疏水性。2-羥基丙基-N-二甲基十四烷基氯化銨改性殼聚糖/SA復合膜對金黃色葡萄球菌的抑菌圈直徑為2.37 cm,對大腸桿菌的抑菌圈直徑為1.75 cm。
實施例12
稱取一定量的取代度為0.60的2-羥基丙基-N-二甲基十六烷基氯化銨改性殼聚糖粉末和羧甲基纖維素鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為5.0的2-羥基丙基-N-二甲基十六烷基氯化銨改性殼聚糖溶液和質量分數為5.0的羧甲基纖維素鈉溶液;
按溶質質量比為3:10的比例稱取上述配置的2-羥基丙基-N-二甲基十六烷基氯化銨改性殼聚糖溶液與羧甲基纖維素鈉溶液,并于20 ℃水浴下混合,制得均一的2-羥基丙基-N-二甲基十六烷基氯化銨改性殼聚糖/CMC水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱42 ℃干燥24 h,待溶劑完全蒸發后,即得到干的2-羥基丙基-N-二甲基十六烷基氯化銨改性殼聚糖/CMC復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的2-羥基丙基-N-二甲基十六烷基氯化銨改性殼聚糖分子和羧甲基纖維素鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為29.53 MPa,比純CMC膜的抗張強度增加了147.1%;斷裂伸長率為10.16%。水在復合膜的接觸角為143o,大于在純CMC膜的接觸角,表現出良好的疏水性。2-羥基丙基-N-二甲基十六烷基氯化銨改性殼聚糖/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為2.57 cm,對大腸桿菌的抑菌圈直徑為1.85 cm。
實施例13
稱取一定量的取代度為0.75的2,3-環氧丙基三甲基氯化銨改性殼聚糖粉末和海藻酸鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為2.5的2,3-環氧丙基三甲基氯化銨改性殼聚糖溶液和質量分數為2.5的海藻酸鈉溶液;
按溶質質量比為0.8:10的比例稱取上述配置的2,3-環氧丙基三甲基氯化銨改性殼聚糖溶液與海藻酸鈉溶液,并于18 ℃水浴下混合,制得均一的2,3-環氧丙基三甲基氯化銨改性殼聚糖/SA水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱40 ℃干燥120 h,待溶劑完全蒸發后,即得到干的2,3-環氧丙基三甲基氯化銨改性殼聚糖/SA復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的2,3-環氧丙基三甲基氯化銨改性殼聚糖分子和海藻酸鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為15.16 MPa,比純SA膜的抗張強度增加了77.10%;斷裂伸長率為2.23%。水在復合膜的接觸角為86o,大于在純SA膜的接觸角,表現出良好的疏水性。2,3-環氧丙基三甲基氯化銨改性殼聚糖/SA復合膜對金黃色葡萄球菌的抑菌圈直徑為1.08 cm,對大腸桿菌的抑菌圈直徑為0.97 cm。
實施例14
稱取一定量的取代度為0.91的O-甲基羧基-3-氯-2-羥丙基三甲基氯化銨改性殼聚糖粉末和羧甲基纖維素鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為3.0的O-甲基羧基-3-氯-2-羥丙基三甲基氯化銨改性殼聚糖溶液和質量分數為3.0的羧甲基纖維素鈉溶液;
按溶質質量比為1.8:10的比例稱取上述配置的O-甲基羧基-3-氯-2-羥丙基三甲基氯化銨改性殼聚糖溶液與羧甲基纖維素鈉溶液,并于30 ℃水浴下混合,制得均一的O-甲基羧基-3-氯-2-羥丙基三甲基氯化銨改性殼聚糖/CMC水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱45 ℃干燥30 h,待溶劑完全蒸發后,即得到干的O-甲基羧基-3-氯-2-羥丙基三甲基氯化銨改性殼聚糖/CMC復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的O-甲基羧基-3-氯-2-羥丙基三甲基氯化銨改性殼聚糖分子和CMC分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為20.63 MPa,比純CMC膜的抗張強度增加了72.64%;斷裂伸長率為6.82%。水在復合膜的接觸角為90o,大于在純CMC膜的接觸角,表現出良好的疏水性。O-甲基羧基-3-氯-2-羥丙基三甲基氯化銨改性殼聚糖/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為2.67 cm,對大腸桿菌的抑菌圈直徑為2.57 cm。
實施例15
稱取一定量的取代度為0.88的O-甲基羧基-2,3-環氧丙基三甲基氯化銨改性殼聚糖粉末和海藻酸鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為4.0的O-甲基羧基-2,3-環氧丙基三甲基氯化銨改性殼聚糖溶液和質量分數為4.0的海藻酸鈉溶液;
按溶質質量比為2.5:10的比例稱取上述配置的O-甲基羧基-2,3-環氧丙基三甲基氯化銨改性殼聚糖溶液與海藻酸鈉溶液,并于55 ℃水浴下混合,制得均一的O-甲基羧基-2,3-環氧丙基三甲基氯化銨改性殼聚糖/SA水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱58 ℃干燥40 h,待溶劑完全蒸發后,即得到干的O-甲基羧基-2,3-環氧丙基三甲基氯化銨改性殼聚糖/SA復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的O-甲基羧基-3-氯-2-羥丙基三甲基氯化銨改性殼聚糖分子和海藻酸鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為18.33 MPa,比純SA膜的抗張強度增加了114.1%;斷裂伸長率為15.36%。水在復合膜的接觸角為97o,大于在純SA膜的接觸角,表現出良好的疏水性。O-甲基羧基-3-氯-2-羥丙基三甲基氯化銨改性殼聚糖/SA復合膜對金黃色葡萄球菌的抑菌圈直徑為2.07 cm,對大腸桿菌的抑菌圈直徑為1.83 cm。
實施例16
稱取一定量的取代度為0.58的O-乙基羧基-2,3-環氧丙基二甲基十四烷基氯化銨改性殼聚糖粉末和海藻酸鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為2.0的O-乙基羧基-2,3-環氧丙基二甲基十四烷基氯化銨改性殼聚糖溶液和質量分數為2.0的海藻酸鈉溶液;
按溶質質量比為2.8:10的比例稱取上述配置的O-乙基羧基-2,3-環氧丙基二甲基十四烷基氯化銨改性殼聚糖溶液與海藻酸鈉溶液,并于25 ℃水浴下混合,制得均一的O-乙基羧基-2,3-環氧丙基二甲基十四烷基氯化銨改性殼聚糖/SA水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱60 ℃干燥70 h,待溶劑完全蒸發后,即得到干的O-乙基羧基-2,3-環氧丙基二甲基十四烷基氯化銨改性殼聚糖/SA復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的O-乙基羧基-2,3-環氧丙基二甲基十四烷基氯化銨改性殼聚糖分子和海藻酸鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為17.33 MPa,比純SA膜的抗張強度增加了102.4%;斷裂伸長率為13.30%。水在復合膜的接觸角為128o,大于在純SA膜的接觸角,表現出良好的疏水性。O-乙基羧基-2,3-環氧丙基二甲基十四烷基氯化銨改性殼聚糖/SA復合膜對金黃色葡萄球菌的抑菌圈直徑為2.37 cm,對大腸桿菌的抑菌圈直徑為2.15 cm。
實施例17
稱取一定量的取代度為0.90的O-丙基羧基-2,3-環氧丙基二甲基十八烷基氯化銨改性殼聚糖粉末和海藻酸鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為1.0的O-丙基羧基-2,3-環氧丙基二甲基十八烷基氯化銨改性殼聚糖溶液和質量分數為1.0的海藻酸鈉溶液;
按溶質質量比為2.2:10的比例稱取上述配置的O-丙基羧基-2,3-環氧丙基二甲基十八烷基氯化銨改性殼聚糖溶液與海藻酸鈉溶液,并于45 ℃水浴下混合,制得均一的O-丙基羧基-2,3-環氧丙基二甲基十八烷基氯化銨改性殼聚糖/SA水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱35 ℃干燥110 h,待溶劑完全蒸發后,即得到干的O-丙基羧基-2,3-環氧丙基二甲基十八烷基氯化銨改性殼聚糖/SA復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的O-丙基羧基-2,3-環氧丙基二甲基十八烷基氯化銨改性殼聚糖分子和海藻酸鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為17.90 MPa,比純SA膜的抗張強度增加了109.1%;斷裂伸長率為14.63%。水在復合膜的接觸角為115o,大于在純SA膜的接觸角,表現出良好的疏水性。O-丙基羧基-2,3-環氧丙基二甲基十八烷基氯化銨改性殼聚糖/SA復合膜對金黃色葡萄球菌的抑菌圈直徑為2.07 cm,對大腸桿菌的抑菌圈直徑為2.05 cm。
實施例18
稱取一定量的取代度為0.85的O-乙基羧基-2,3-環氧丙基二甲基癸基氯化銨改性殼聚糖粉末和羧甲基纖維素鈉粉末,分別向其中加入一定量的去離子水,先于60 ℃水浴下恒溫溶脹一定時間,然后攪拌至均勻,攪拌結束后于室溫下放置24 h,最終分別得到質量分數為4.5的O-乙基羧基-2,3-環氧丙基二甲基癸基氯化銨改性殼聚糖溶液和質量分數為4.5的羧甲基纖維素鈉溶液;
按溶質質量比為2.7:10的比例稱取上述配置的O-乙基羧基-2,3-環氧丙基二甲基癸基氯化銨改性殼聚糖溶液與羧甲基纖維素鈉溶液,并于58 ℃水浴下混合,制得均一的O-乙基羧基-2,3-環氧丙基二甲基癸基氯化銨改性殼聚糖/CMC水溶液,然后將混合溶液倒入膜具,使混合溶液的厚度盡可能均勻,放入鼓風干燥箱50 ℃干燥60 h,待溶劑完全蒸發后,即得到干的O-乙基羧基-2,3-環氧丙基二甲基癸基氯化銨改性殼聚糖/CMC復合膜。
本實施例制備的復合膜具有良好的透光性,膜的切面和傅里葉變化紅外譜圖顯示復合膜中的O-乙基羧基-2,3-環氧丙基二甲基癸基氯化銨改性殼聚糖分子和羧甲基纖維素鈉分子之間存在良好的相容性和顯著的相互作用;復合膜的抗張強度為27.90 MPa,比純CMC膜的抗張強度增加了133.5%;斷裂伸長率為15.06%。水在復合膜的接觸角為120o,大于在純CMC膜的接觸角,表現出良好的疏水性。O-乙基羧基-2,3-環氧丙基二甲基癸基氯化銨改性殼聚糖/CMC復合膜對金黃色葡萄球菌的抑菌圈直徑為2.10 cm,對大腸桿菌的抑菌圈直徑為2.15 cm。
以上所述,僅用于說明本發明,而不應視為限定本發明的范圍,任何熟悉本專業的技術人員可能利用上述揭示的技術內容加以變更或改型為等同變化的等效實施例。例如,具體地用2,3-環氧丙基二甲基十八烷基氯化銨、2,3-環氧丙基二甲基癸基氯化銨、2,3-環氧丙基二甲基十二烷基氯化銨、2,3-環氧丙基二甲基十四烷基氯化銨、2,3-環氧丙基二甲基十六烷基氯化銨或3-氯-2-羥丙基三甲基氯化銨中的一種替換2,3-環氧丙基三甲基氯化銨,用O-甲基羧基改性殼聚糖、O-乙基羧基改性殼聚糖或O-丙基羧基改性殼聚糖中的一種替換殼聚糖,用1-烯丙基-3-甲基氯化咪唑、1-丁基-3-甲基氯化咪唑或1-乙基-3-甲基咪唑醋酸鹽中的一種替換1-丁基-3-甲基溴化咪唑,仍按照上述制備改性殼聚糖的方法,在不同條件下制備出不同取代度的相應的改性殼聚糖季銨鹽。但是凡是未脫離本發明技術方案內容,依據本發明的技術實質對以上實施例所作的任何簡單修改、等同變化與改型,仍屬于本發明技術方案的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!