一種黃體酮雜質的合成方法與流程
本發明涉及有機合成領域,具體涉及一種特定黃體酮雜質(產物ⅱ)的合成方法,解決該雜質在現有技術中合成困難、收率低、成本高以及難以獲得純凈對照品等問題,為黃體酮原料藥的質量控制和相關研究提供支持。
背景技術:
1、黃體酮作為一種具有重要生理活性的甾體激素,在婦科疾病治療(如調節月經周期、維持妊娠等)以及輔助生殖技術領域發揮著不可或缺的作用。其臨床應用廣泛,使得對黃體酮原料藥的質量要求更為嚴格。黃體酮原料藥的化學結構的20位上會產生一種r構型的雜質(即產物ⅱ)。這一雜質的存在,不僅影響原料藥的純度和質量穩定性,還對藥物的療效和安全性產生潛在威脅。雜質含量過高改變藥物的藥代動力學特性,影響藥物在體內的吸收、分布、代謝和排泄過程,進而降低治療效果或引發不良反應。
2、然而,該雜質特殊的構型翻轉現象導致其合成路線復雜,合成難度極大。傳統的合成方法難以精準控制反應條件,使得反應過程中副反應增多,目標產物收率降低,不僅造成了原料的浪費,增加了生產成本,還使得產品質量難以達到理想標準。同時,在獲取純凈的該雜質對照品方面,傳統的原料藥雜質分離純化方法也有缺陷。常用的結晶、萃取、色譜等分離純化技術,在處理這種構型特殊且含量較低的雜質時,面臨著選擇性差、分離效率低等問題。即使經過復雜的操作流程,也很難獲得高純度的產物ⅱ對照品,這嚴重制約了對該雜質的深入研究和準確檢測,無法為原料藥的質量控制提供可靠的依據。
3、此前,已有相關專利對產物ⅱ的合成方法進行了報道。專利ep3275888a1公開的合成途徑中,第一步選用四氫鋁鋰作為還原劑對黃體酮進行還原反應,該試劑具有較強的還原性,但反應條件較為苛刻,需要嚴格控制反應溫度、溶劑等條件,否則容易引發過度還原等副反應。隨后使用新鮮制備的二氧化錳進行選擇性氧化,二氧化錳的氧化選擇性雖然相對較高,但在實際操作中,其氧化效果受多種因素影響,如二氧化錳的活性、用量、反應時間等。最終得到的產物ⅰ和產物ⅱ的比例僅為1:0.1,這意味著在合成過程中,大量的原料轉化為產物ⅰ,而目標產物ⅱ的產量極低。若要滿足研究或生產對產物ⅱ的需求,就必須投入遠超預期的物料成本,這對于大規模生產來說是不經濟的,也限制了該方法在實際生產中的應用。
4、專利cn113788871a所報道的合成方法同樣致力于解決產物ⅱ的合成問題。其第一步采用原乙酸三乙酯對黃體酮3位酮基進行保護,這一步驟旨在通過保護基團的引入來調控反應活性和選擇性。第二步使用硼氫化鈉進行還原反應,硼氫化鈉是一種常用的還原劑,具有反應條件溫和、選擇性較好等優點。第三步則使用無機酸進行脫保護反應,以去除之前引入的保護基團,得到目標產物。然而,盡管該方法在一定程度上優化了反應步驟,但最終產物ⅰ和產物ⅱ的比例約為1:0.4,雖然較專利ep3275888a1有所提高,但仍然不盡人意。產物ⅱ的相對低收率依然導致合成成本居高不下,且難以滿足市場對高純度產物ⅱ的需求。現有的合成方法在產物ⅱ的合成上均存在不同程度的缺陷,無法實現高效、低成本的合成目標。
技術實現思路
1、本發明的目的是提供一種黃體酮雜質(產物ⅱ)的合成方法,克服現有技術中合成困難、收率低、成本高的問題,提高產物ⅱ的合成效率。
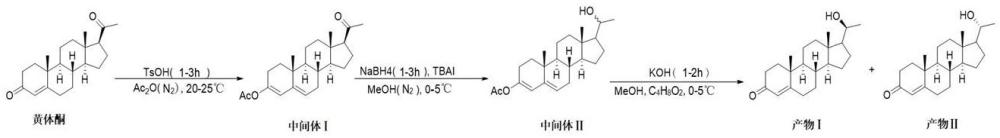
2、本發明提供的一種黃體酮雜質合成方法,通過明確且有序的步驟,包括中間體ⅰ的合成(涉及原料混合、氮氣置換、催化反應、淬滅沉淀、過濾純化等操作)、中間體ⅱ的合成(在低溫和氮氣保護下進行原料混合、分三批加還原劑、淬滅、萃取、干燥濃縮)以及產物ⅰ和產物ⅱ的合成(從低溫投料混合、堿液反應、萃取、干燥濃縮到高壓制備分離),精確控制了反應條件和過程,不僅能夠有效合成特定比例(1:0.73-0.75)的產物ⅰ和產物ⅱ,提高了目標雜質(產物ⅱ)的收率,解決了現有技術中合成困難、收率低、成本高以及難以獲得純凈對照品的問題,而且為黃體酮原料藥的質量控制和相關研究提供了有力支持,確保了合成過程的可重復性和產物的高質量。
3、本發明提供一種黃體酮雜質的合成方法,具體包括以下步驟:
4、s1中間體ⅰ的合成步驟在室溫下將黃體酮加入醋酸酐后進行氮氣置換,排除反應體系中的氧氣,防止反應物或產物被氧化,創造惰性反應環境,有利于后續反應的順利進行;對甲苯磺酸作為催化劑加入,有效降低反應活化能,加快反應速率,促進黃體酮3位酮基與醋酸酐的反應,提高反應效率,使反應在合理時間內達到較高的轉化率;隨后通過倒入冰水、攪拌、過濾、打漿、再過濾和干燥等操作,逐步分離和純化中間體ⅰ,具體包括反應結束后倒入冰水,降低產物溶解度,使中間體ⅰ以固體形式析出,便于與反應體系中的其他物質分離,初步純化中間體ⅰ,過濾收集濾餅,然后用甲醇打漿,甲醇可溶解部分雜質,進一步純化中間體ⅰ,再次過濾得到更純凈的中間體ⅰ濾餅,最后干燥去除殘留溶劑,得到高純度的白色固體中間體ⅰ,為下一步反應提供高質量原料。
5、因此,在室溫條件下,將黃體酮原料加入到醋酸酐中,對反應體系進行氮氣置換,在攪拌狀態下加入對甲苯磺酸于室溫繼續攪拌1-3小時,將反應液倒入冰水中,析出固體,攪拌10-30分鐘后進行過濾操作,收集濾餅,將濾餅加入甲醇中打漿0.5-1.5小時,再次過濾,干燥得到白色固體即為中間體ⅰ。
6、s2中間體ⅱ的合成步驟冰浴條件下將中間體ⅰ和四丁基碘化銨加入甲醇并氮氣置換,冰浴控制反應溫度,降低副反應發生概率,氮氣置換保持反應體系惰性,四丁基碘化銨起到相轉移催化等作用,促進后續硼氫化鈉還原反應的進行,提高反應選擇性和收率;分三批加入硼氫化鈉并保溫攪拌,避免反應過于劇烈,使硼氫化鈉與中間體ⅰ充分反應,將特定官能團還原,得到中間體ⅱ,這種加入方式有助于控制反應速率和選擇性,提高目標產物的生成量;反應結束后的淬滅、萃取、干燥和濃縮等操作,可有效分離得到中間體ⅱ,具體包括反應結束后用飽和氯化銨淬滅未反應的硼氫化鈉,調節反應體系ph值,使反應停止,防止過度反應,同時避免硼氫化鈉殘留對后續操作和產物質量產生影響;乙酸乙酯萃取將中間體ⅱ轉移至有機相,與水相中的雜質分離,無水硫酸鈉干燥去除有機相中的殘留水分,減壓濃縮得到白色固體中間體ⅱ,實現了中間體ⅱ的有效分離和純化,為最終產物的合成做好準備。
7、因此,在冰浴條件下,將白色固體中間體ⅰ和四丁基碘化銨加入到甲醇中,同樣進行氮氣置換,在攪拌下分三批加入硼氫化鈉,加完后保溫攪拌1-3小時,反應結束后,用飽和氯化銨溶液淬滅反應,終止反應進行后加入乙酸乙酯進行萃取操作,將兩次萃取得到的有機相合并后進行干燥處理,濃縮得到白色固體即為中間體ⅱ。
8、s3產物ⅰ和產物ⅱ的合成步驟在冰浴條件下將中間體ⅱ加入甲醇后加入氫氧化鉀水溶液,低溫控制反應速率,使反應平穩進行,氫氧化鉀水溶液參與反應,促進特定化學鍵的斷裂和形成,生成產物ⅰ和產物ⅱ的混合物,為后續分離提供基礎;隨后升至室溫攪拌1-2小時確保反應完全,加入乙酸乙酯萃取使產物轉移至有機相,合并有機相并干燥濃縮得到粗品,該過程將產物從反應體系中分離出來,同時去除部分雜質,得到相對富集產物的粗品;通過高壓制備色譜對粗品進行分離,利用反相c18柱和特定流動相,在精確控制的泵流速、壓力和柱溫條件下,根據產物ⅰ和產物ⅱ在色譜柱上不同的保留時間進行分離,實現兩者的精確分離純化,得到高質量的白色固體產物ⅰ和產物ⅱ,且控制產物ⅰ和產物ⅱ的比例為1:0.73-0.75,滿足對該雜質合成的要求,不同溫度下的反應控制和多種分離手段的結合,保證了產物的純度和收率。
9、因此,在冰浴條件下,將白色固體中間體ⅱ加入到甲醇中,攪拌狀態下加入氫氧化鉀水溶液,隨后將反應體系升至室溫并攪拌1-2小時,反應完成后,加入乙酸乙酯進行萃取,合并有機相并干燥濃縮,得到粗品;通過高壓制備的方法對粗品進行分離,最終分別得到白色固體產物ⅰ和產物ⅱ,且產物ⅰ和產物ⅱ的比例為1:0.73-0.75;
10、反應式如下:
11、。
12、本發明提供的黃體酮雜質的合成方法中步驟s1中間體ⅰ合成,通過詳細規定各操作環節,在室溫下精確投料并進行氮氣置換,為反應提供穩定環境;以特定速度攪拌下加入適量催化劑并控制反應時間,確保中間體ⅰ有效生成;反應結束后,嚴格控制淬滅與沉淀條件、過濾與初步純化、打漿純化以及再次過濾與干燥,使得中間體ⅰ的合成過程更加精確、可控,有效提高中間體ⅰ的純度和收率,為后續中間體ⅱ及產物的合成提供高質量的原料,保證整個合成路線的順利進行,對提高整體合成工藝的穩定性和產物質量起到關鍵作用。
13、作為優選,所述步驟s1中間體ⅰ的合成具體包括以下操作:
14、s11投料與氮氣置換步驟在室溫環境下,按照60-80g的范圍加入黃體酮(對應物質的量190-255mmol)到300-500ml醋酸酐中,確保反應物的量在合適比例,為后續反應提供充足的原料,保證反應能夠順利進行且達到一定的規模;連接氮氣裝置對反應容器進行置換,排除容器內的空氣,主要是氧氣,氧氣會與反應物或反應中間體發生氧化反應,影響反應的選擇性和收率,氮氣置換后形成的惰性環境有利于保護反應物,使反應按預期進行,得到黃體酮與醋酸酐的混合溶液。
15、s12催化劑加入與反應啟動步驟在200-300rpm的持續攪拌下,將20-30g對甲苯磺酸(物質的量116-175mmol)緩慢加入到混合溶液中,使催化劑均勻分散在反應體系中,避免局部濃度過高引發副反應,同時攪拌有助于催化劑與反應物充分接觸,提高催化效率;室溫下繼續攪拌反應1-3小時,為反應提供足夠的時間,使催化劑能夠充分發揮作用,促進黃體酮與醋酸酐在其催化下發生反應,生成中間體ⅰ,合適的反應時間和溫度控制能夠保證反應的轉化率和選擇性,避免反應不完全或過度反應。
16、s13淬滅與沉淀步驟在反應結束后將反應液緩慢倒入800-1200ml冰水中,冰水的低溫環境使反應迅速停止,防止反應繼續進行產生副產物,同時大量的水稀釋了反應體系,改變了反應的化學平衡,有利于產物的析出;攪拌速度控制在300-400rpm,使反應液與冰水充分混合,一方面確保反應淬滅徹底,另一方面促使產物以沉淀形式析出;快速攪拌增加產物分子之間的碰撞機會,使其更容易聚集形成沉淀,便于后續分離;繼續攪拌10-30分鐘進一步保證沉淀完全,得到中間體ⅰ沉淀。
17、s14過濾與初步純化步驟采用布氏漏斗、濾紙等過濾裝置進行操作,有效分離出沉淀(中間體ⅰ)和溶液,收集到含有中間體ⅰ以及少量雜質的第一濾餅,這種過濾方式簡單高效,可去除大部分反應體系中的液體成分,初步富集中間體ⅰ;雖然得到的濾餅中仍含有少量雜質,但通過這一步驟已經實現了中間體ⅰ與反應體系中大部分可溶性雜質的分離,為后續進一步純化提供了基礎,減少了后續純化的難度和工作量。
18、s15打漿純化步驟將第一濾餅轉移至另一容器中,加入400-600ml甲醇并在250-350rpm攪拌下打漿0.5-1.5小時,甲醇可以溶解濾餅中的部分雜質,而中間體ⅰ在甲醇中的溶解度相對較小,通過這種選擇性溶解作用,進一步去除雜質,提高中間體ⅰ的純度;打漿過程是一種物理純化手段,不涉及化學反應,通過攪拌使甲醇與濾餅充分接觸,雜質溶解在甲醇中,然后通過后續過濾操作可將溶解有雜質的甲醇與純化后的中間體ⅰ分離,達到進一步純化中間體ⅰ的目的。
19、s16再次過濾與干燥步驟在打漿結束后再次過濾,將經過甲醇打漿純化后的中間體ⅰ與含有雜質的甲醇溶液徹底分離,收集得到更為純凈的中間體ⅰ第二濾餅,進一步提高了中間體ⅰ的純度,去除了在打漿過程中未完全分離的雜質;將第二濾餅在40-50℃的真空條件下干燥2-3小時,去除殘留的甲醇溶劑,殘留溶劑會影響中間體ⅰ的穩定性和后續反應的進行,干燥過程確保得到純凈的中間體ⅰ白色固體,使其能夠穩定保存并適合作為下一步反應的原料,保證了整個合成路線的連續性和穩定性,得到純凈的中間體ⅰ白色固體,反應式如下:
20、。
21、在本發明中間體ⅰ和中間體ⅱ的合成過程中,氮氣置換操作的目的是防止原料氧化,減少副反應發生,維持反應選擇性,保證反應環境穩定性,保護催化劑活性,為合成過程提供了靈活且有效的手段來創造惰性反應環境。
22、黃體酮等有機化合物在有氧氣存在的情況下會發生氧化反應,導致原料變質,影響反應的進行和產物的質量,而氮氣是一種惰性氣體,在反應體系中通入氮氣可以排除氧氣,創造一個無氧或低氧的環境,從而保護原料不被氧化,避免當原料發生氧化時,生成其他副產物,降低目標中間體ⅰ和中間體ⅱ的產率,同時引入雜質,增加后續純化的難度,影響整個合成過程的效率和經濟性。許多化學反應在有氧條件下會引發一系列復雜的副反應,氧氣會與反應中間體或活性物種發生反應,生成不需要的副產物,而通過氮氣置換去除氧氣,可以抑制這些潛在的副反應途徑,有利于生成中間體ⅰ和中間體ⅱ;減少副反應意味著更多的原料能夠轉化為目標產物中間體ⅰ和中間體ⅱ,提高了反應的原子經濟性和原料利用率,降低了生產成本,同時也簡化了后續產物分離和純化的步驟。氧化反應的發生會改變反應的路徑,使反應朝著非預期的方向進行,降低目標產物的選擇性,在中間體ⅱ的合成中,涉及到硼氫化鈉等還原劑的使用,若有氧氣存在,則會優先與還原劑反應,消耗還原劑,從而影響對目標中間體ⅱ的合成,使反應選擇性變差,氮氣置換可避免這種情況,提高反應生成目標中間體的選擇性。氮氣的化學性質穩定,在反應過程中不參與化學反應,能夠維持反應體系的穩定性,可以防止反應體系受到水分、二氧化碳等外界空氣成分的干擾,確保反應在一個相對恒定的環境中進行,而穩定的反應環境對于實驗的重現性至關重要。對甲苯磺酸作為反應的催化劑,其活性會受到氧氣或其他雜質氣體的影響,在無氧的氮氣環境下,可以減少催化劑失活的可能性,延長催化劑的使用壽命,保證其在反應過程中持續有效地催化反應進行,從而穩定反應速率和產物收率;保持催化劑的活性不僅有助于提高當前反應的效率,減少催化劑的用量,降低生產成本,而且還可以避免因催化劑失活而導致的反應失敗或重新制備催化劑的麻煩,提高整個合成工藝的經濟性和可靠性。
23、作為優選,所述氮氣置換操作采用的方式包括:直接通氣法、多次通氣-排空法、真空-氮氣填充法。所述直接通氣法操作,能夠在相對較短的時間內,用氮氣替代大部分空氣,操作簡單直接,可有效減少反應容器內氧氣等空氣成分的含量,為反應提供一個相對惰性的環境;具體包括:將氮氣鋼瓶作為氮氣源通過管道連接至反應容器的進氣口,打開氮氣鋼瓶閥門,調節氮氣流量至100-300ml/min,持續通入氮氣5-15分鐘,使氮氣充滿反應容器,將容器內的空氣從排氣口排出,關閉氮氣鋼瓶閥門,停止通氣,完成一次氮氣置換。所述多次通氣-排空法操作相比直接通氣法,進一步降低氧氣殘留量,對于對氧氣較為敏感的反應,能提供更好的保護,減少因微量氧氣存在而引發的副反應,每次循環都能進一步稀釋容器內的空氣,確保容器內空氣被充分置換,為反應創造更嚴格的惰性條件,提高反應的選擇性和收率;具體包括:連接氮氣源與反應容器進氣口,打開氮氣鋼瓶閥門,調節流量至100-300ml/min,向反應容器內通入氮氣1-2分鐘,關閉氮氣鋼瓶閥門,同時打開反應容器的排氣口,將容器內的氣體排空,排空時間控制在30秒-1分鐘,重復上述通氣-排空步驟3-5次,確保容器內空氣被充分置換。所述真空-氮氣填充法操作在負壓作用下,容器內的空氣(包括氧氣、氮氣、二氧化碳等)被抽出,極大程度地降低了空氣成分的含量,幾乎可以去除所有可揮發性雜質氣體,為后續填充氮氣創造了極為純凈的空間,對于涉及高活性中間體或催化劑的反應,能有效防止反應過程中的氧化、水解等副反應,確保反應的順利進行和產物的高質量;具體包括:使用真空泵連接反應容器,將容器內抽至-0.08mpa至-0.1mpa的真空狀態,保持1-2分鐘,使容器內的空氣被抽出,關閉真空泵,然后將氮氣源連接至反應容器進氣口,緩慢打開氮氣鋼瓶閥門,向容器內填充氮氣,直至容器內壓力恢復至常壓,重復上述真空-氮氣填充步驟2-3次,完成氮氣置換操作。
24、本發明提供的黃體酮雜質的合成方法中步驟s2中間體ⅱ合成,通過在0-5℃冰浴及氮氣保護下精確混合原料,為反應營造穩定且惰性的環境,抑制副反應,提高反應選擇性;以特定方式加入還原劑,確保反應平穩、充分進行,生成中間體ⅱ;用飽和氯化銨溶液淬滅反應并調節ph值,終止反應并穩定體系;經兩次乙酸乙酯萃取、無水硫酸鈉干燥和減壓濃縮等操作,有效分離、純化中間體ⅱ,提高其純度和收率,為后續產物合成提供高質量前體,對整個合成工藝的高效推進和產物質量保障至關重要。
25、作為優選,所述步驟s2中間體ⅱ的合成具體包括以下操作:
26、s21低溫混合與氮氣保護:在0-5℃冰浴條件下,將60-80g中間體ⅰ(物質的量118-225mmol)和5-10g四丁基碘化銨(物質的量13-27mmol)加入到500-800ml甲醇中,置于反應容器內,連接氮氣裝置,對反應容器進行氮氣置換操作,采用多次通氣-排空法或真空-氮氣填充法,確保容器內空氣被氮氣完全置換,得到中間體ⅰ、四丁基碘化銨與甲醇的混合溶液;在0-5℃冰浴條件下進行操作,降低反應體系的溫度,低溫有助于抑制硼氫化鈉等還原劑的副反應,提高反應的選擇性,有利于目標中間體ⅱ的生成;連接氮氣裝置并采用多次通氣-排空法或真空-氮氣填充法進行氮氣置換,確保容器內空氣被完全置換,防止氧氣等氣體與反應物或反應中間體發生氧化反應,同時排除其他干擾反應的氣體(如二氧化碳、水蒸氣等),在惰性氮氣環境下,中間體ⅰ和四丁基碘化銨能夠更穩定地存在,保證反應按預期進行,提高反應的收率和產物的純度;將60-80g中間體ⅰ和5-10g四丁基碘化銨加入到500-800ml甲醇中,甲醇作為溶劑,在低溫和攪拌下有助于原料的溶解和均勻混合,均勻的反應體系有利于反應物分子之間的有效碰撞,為后續反應的順利進行提供良好的基礎,使反應能夠在整個體系中均勻地發生。
27、s22還原劑加入與反應進行:在150-250rpm持續攪拌下,將8-12g硼氫化鈉分三批緩慢加入到上述混合溶液中,保持冰浴溫度,持續攪拌反應1-3小時,使反應充分進行,生成中間體ⅱ;在150-250rpm持續攪拌下,將8-12g硼氫化鈉分三批緩慢加入到混合溶液中,這種緩慢分批加入的方式避免硼氫化鈉一次性加入導致反應過于劇烈,使反應熱能夠及時散發,有利于控制反應速率,同時,通過分批加入更好地調節反應的進程,提高反應的選擇性,使硼氫化鈉優先與目標官能團發生反應,減少對其他基團的不必要還原,從而提高中間體ⅱ的生成效率;保持冰浴溫度并持續攪拌反應1-3小時,持續攪拌保證了反應物的充分接觸,使反應均勻進行,在低溫下進行反應,結合前面的氮氣保護和緩慢加樣措施,進一步抑制了副反應的發生,使硼氫化鈉能夠按照預期對中間體ⅰ進行還原反應,生成中間體ⅱ,確保反應的選擇性和收率。
28、s23淬滅反應:反應結束后,將500-800ml飽和氯化銨溶液緩慢加入到反應容器中,淬滅未反應的硼氫化鈉并調節反應體系的ph值,加入過程中不斷攪拌,攪拌速度控制在200-300rpm;反應結束后,將500-800ml飽和氯化銨溶液緩慢加入到反應容器中,飽和氯化銨溶液中的銨離子與未反應的硼氫化鈉發生反應,使其失去還原活性,從而終止反應,避免硼氫化鈉未被完全淬滅而在后續處理過程中繼續反應,導致產物不穩定或產生其他雜質;同時,飽和氯化銨溶液呈酸性,加入后可以調節反應體系的ph值,合適的ph值對于后續的萃取操作以及產物的穩定性有重要影響,通過加入飽和氯化銨溶液調節ph值,避免產物在過酸或過堿的環境下發生水解或其他化學變化,使反應體系處于一個相對穩定的狀態,有利于后續步驟的進行。
29、s24萃取操作:淬滅完成后,將500-800ml乙酸乙酯加入反應容器中,充分振蕩并攪拌,攪拌速度控制在200-300rpm,使產物轉移至乙酸乙酯相中;靜置分層后,將有機相即乙酸乙酯相轉移至分液漏斗中收集,將水相再用500-800ml乙酸乙酯重復萃取一次,合并兩次萃取得到的有機相;淬滅完成后,加入500-800ml乙酸乙酯并充分振蕩攪拌,乙酸乙酯作為有機溶劑,能夠將產物(中間體ⅱ)從水相轉移到有機相中,這是基于產物在乙酸乙酯和水相中的溶解度差異,通過萃取實現了產物與水相中大部分水溶性雜質(如無機鹽、未反應完的極性試劑等)的分離,初步純化了產物;靜置分層后,將有機相收集,水相再用500-800ml乙酸乙酯重復萃取一次,多次萃取可以進一步提高產物的收率,將殘留在水相中的產物提取到有機相中,減少產物的損失,使更多的中間體ⅱ被富集到有機相中,為后續的干燥和濃縮操作提供更純凈、更富集產物的溶液。
30、s25干燥與濃縮:在有機相中加入無水硫酸鈉作為干燥劑,攪拌10-20分鐘除去殘留的水分,過濾除去干燥劑,得到干燥的有機相,將有機相在減壓條件下進行濃縮,溫度控制在30-40℃,壓力在-0.08--0.1mpa,直至得到白色固體中間體ⅱ;在有機相中加入無水硫酸鈉作為干燥劑,攪拌10-20分鐘,無水硫酸鈉與有機相中殘留的微量水分結合,形成結晶水合物,從而除去水分,水分的存在會影響后續濃縮過程以及產物的穩定性,干燥操作確保有機相的干燥,為下一步濃縮做好準備;過濾除去干燥劑后,將有機相在減壓條件下濃縮,溫度控制在30-40℃,壓力在-0.08--0.1mpa,減壓濃縮在較低溫度下快速蒸發溶劑(乙酸乙酯),避免高溫對產物造成的破壞(如分解、氧化等),隨著溶劑的逐漸蒸發,中間體ⅱ從溶液中析出,最終得到白色固體中間體ⅱ,完成了中間體ⅱ的合成和初步純化,得到的中間體ⅱ作為下一步反應(合成產物ⅰ和產物ⅱ)的原料,保證了整個合成路線的連續性,得到的中間體ⅱ反應式如下:
31、。
32、分三批加入硼氫化鈉的操作方式,整體上是為了實現對反應的精細控制,避免反應的劇烈程度難以控制而引發副反應,確保中間體ⅱ的合成過程平穩、高效地進行,提高產物的質量和反應的選擇性;同時,這種操作方式有助于控制反應的速率和進程,在整個反應過程中,通過多次分批添加和充分攪拌,最大程度地提高硼氫化鈉的利用效率,保證反應朝著生成中間體ⅱ的方向進行,為后續產物ⅰ和產物ⅱ的合成提供高質量的中間體ⅱ原料。
33、進一步地,所述步驟s2中間體ⅱ的合成中硼氫化鈉分三批加入具體包括以下操作:
34、第一批加入:取硼氫化鈉總量的1/3,即2.7-4g硼氫化鈉(物質的量70-105mmol),緩慢加入到中間體ⅰ、四丁基碘化銨與甲醇的混合溶液中,加入過程中持續攪拌,攪拌速度為150-250rpm,加入后繼續攪拌5-10分鐘;緩慢加入避免反應一開始就過于劇烈,防止反應熱瞬間釋放過多,避免局部過熱引發副反應,確保反應在相對溫和的條件下開始;加入后繼續攪拌,使加入的硼氫化鈉能夠與中間體ⅰ和四丁基碘化銨開始發生反應,啟動整個還原反應過程,同時攪拌使反應物充分混合,提高反應的均勻性和反應效率,為后續的反應進程奠定基礎。
35、第二批加入:取硼氫化鈉總量的1/3,按照第一批加入的方式,緩慢加入到反應體系中,持續攪拌,控制加入速度,加入后攪拌5-10分鐘;分批加入的操作進一步推動反應的進行,在第一批加入的基礎上,通過分批次操作,避免反應過于劇烈,使反應平穩進行,保持反應的可操控性;攪拌和控制加入速度確保反應體系的均勻性和穩定性,加入后繼續攪拌,為反應提供足夠的時間讓第二批加入的硼氫化鈉與反應物充分反應,有助于維持反應體系中反應物的濃度梯度,避免反應過于劇烈或不均勻,進一步提高反應的選擇性。
36、第三批加入:將剩余的2.7-4g硼氫化鈉全部緩慢加入并攪拌,加入后持續攪拌反應1-2小時,完成整個硼氫化鈉的加入和反應過程;此時,前兩批硼氫化鈉已部分消耗反應物,第三批的加入可以確保反應體系中有足夠的還原劑將中間體ⅰ完全地還原為中間體ⅱ,保證反應充分進行,提高產物的轉化率和收率;通過持續攪拌,使最后一批硼氫化鈉與反應體系充分接觸,確保反應完全,使中間體ⅰ得到充分的還原,同時較長的攪拌時間為反應達到平衡和中間體ⅱ的充分生成提供保障,減少未反應的中間體ⅰ殘留,有利于提高中間體ⅱ的產率和純度。
37、本發明提供的黃體酮雜質的合成方法中步驟s3產物ⅰ和產物ⅱ合成,從低溫,下精確投料并混合開始,為反應提供適宜的起始條件,利于中間體ⅱ溶解;緩慢加入堿液并經室溫攪拌1-2小時,控制反應啟動和進程,確保反應充分進行生成產物混合物;通過兩次乙酸乙酯萃取、無水硫酸鈉干燥和減壓濃縮,有效分離和富集產物得到粗產物;最后利用高壓制備色譜在特定條件(特定流動相、柱溫、流速和壓力)下進行分離純化,結合餾分收集、旋轉蒸發濃縮和真空干燥,實現產物ⅰ和產物ⅱ的精確分離和高質量制備,得到特定比例(1:0.73-0.75)的產物,為后續研究或質量控制提供高純度的樣品,對整個合成工藝的成功完成和產物質量的精準控制具有關鍵意義。
38、作為優選,所述步驟s3產物ⅰ和產物ⅱ的合成具體包括以下操作:
39、s31低溫投料與混合:在冰浴條件下,將50-70g中間體ⅱ(物質的量139-196mmol)加入到500-700ml甲醇中,置于反應容器內,開啟攪拌裝置,將攪拌速度控制在150-250rpm,使中間體ⅱ充分溶解,得到中間體ⅱ的甲醇溶液;在冰浴條件下,將50-70g中間體ⅱ加入到500-700ml甲醇中并攪拌,低溫有助于中間體ⅱ在甲醇中的溶解,中間體ⅱ在低溫下具有更好的溶解性或穩定性,避免在溶解過程中發生不必要的化學反應或分解,保證其以分子狀態均勻分散在溶劑中,為后續反應提供均一的反應體系;攪拌速度控制在150-250rpm,合適的攪拌速度加速中間體ⅱ的溶解過程,同時避免因攪拌過快引入過多空氣或造成溶液飛濺等問題,通過低溫投料與攪拌混合,使中間體ⅱ充分溶解形成甲醇溶液,為下一步堿液加入和反應啟動做好準備,確保反應從一個穩定、均勻的狀態開始。
40、s32堿液加入與反應啟動:在攪拌狀態下,將配制好的25-35g氫氧化鉀水溶液(物質的量446-626mmol)緩慢加入到上述中間體ⅱ的甲醇溶液中。加完堿液后,將反應容器從冰浴中取出,放置于室溫下,繼續攪拌反應1-2小時,使反應充分進行,生成產物ⅰ和產物ⅱ的混合物;在攪拌狀態下緩慢加入25-35g氫氧化鉀水溶液,緩慢加入防止氫氧化鉀與中間體ⅱ瞬間反應過于劇烈,避免局部濃度過高導致副反應增加,氫氧化鉀在反應中起到關鍵作用,其緩慢加入使反應能夠平穩啟動,有利于控制反應速率,使反應按照預期的方向進行;加完堿液后將反應容器從冰浴取出置于室溫下繼續攪拌1-2小時,升溫過程使反應體系的能量增加,分子運動加快,促進反應物分子之間的有效碰撞,加快反應速率,確保反應充分進行,使中間體ⅱ在氫氧化鉀的作用下發生反應,生成產物ⅰ和產物ⅱ的混合物,提高產物的轉化率。
41、s33萃取操作:反應結束后,向反應容器中加入500-700ml乙酸乙酯,充分振蕩并攪拌,攪拌速度控制在200-300rpm,促使產物轉移至乙酸乙酯相中,靜置分層后,將有機相轉移至分液漏斗中收集,水相再用500-700ml乙酸乙酯重復萃取一次,合并兩次萃取得到的有機相;反應結束后加入500-700ml乙酸乙酯并振蕩攪拌,乙酸乙酯作為有機溶劑,與水相不互溶,利用產物在乙酸乙酯和水相中的溶解度差異,將產物(產物ⅰ和產物ⅱ)從水相萃取到有機相中,實現產物與水相中水溶性雜質(如未反應完的氫氧化鉀、反應生成的鹽等)的初步分離,提高產物的純度;靜置分層后收集有機相,水相再用500-700ml乙酸乙酯重復萃取一次,多次萃取可進一步提高產物的萃取效率,減少產物在水相中的殘留,增加產物的收率,使更多的產物被富集到有機相中,為后續的干燥和濃縮操作提供更純凈、更富集產物的溶液。
42、s34干燥與濃縮:在有機相中加入無水硫酸鈉作為干燥劑,攪拌10-20分鐘除去殘留的水分,過濾除去干燥劑,得到干燥的有機相,將有機相在減壓條件下進行濃縮,控制溫度在30-40℃,壓力在-0.08--0.1mpa,得到粗產物;在有機相中加入無水硫酸鈉攪拌10-20分鐘,無水硫酸鈉吸收有機相中殘留的微量水分,形成結晶水合物,從而除去水分,水分的存在會影響后續濃縮過程以及產物的穩定性,干燥操作確保有機相干燥,防止在濃縮過程中因水分存在而發生水解等副反應,保證產物質量;過濾除去干燥劑后,在減壓條件下濃縮有機相,溫度控制在30-40℃,壓力在-0.08--0.1mpa,減壓濃縮在較低溫度下快速蒸發乙酸乙酯溶劑,避免高溫對產物造成破壞(如分解、氧化等),隨著溶劑的蒸發,產物濃度逐漸增加,最終得到粗產物,完成了從反應混合物到相對富集產物的粗品的轉變,為后續高壓制備分離提供原料,減少雜質對高壓制備分離過程的干擾。
43、s35高壓制備分離:將粗產物用少量甲醇或乙腈-水的流動相溶解并經0.22-0.45μm微孔濾膜過濾配制成樣品溶液,選用配備反相c18柱,以乙腈-水為流動相,在泵流速0.8-1.2ml/min、壓力10-20mpa、柱溫30-40℃條件下,通過高壓制備色譜進行分離純化,利用檢測器記錄色譜峰并依出峰時間用餾分收集器收集含產物ⅰ和產物ⅱ的洗脫液餾分,將餾分轉移至圓底燒瓶,用旋轉蒸發儀在30-40℃水浴、-0.08--0.1mpa壓力下減壓濃縮得到白色固體,最后將其置于真空干燥箱中,在40-50℃、-0.08--0.1mpa真空度下干燥2-4小時,獲得高質量的產物ⅰ和產物ⅱ,且產物ⅰ和產物ⅱ的比例為1:0.73-0.75;用少量甲醇或乙腈-水的流動相溶解粗產物并過濾,得到純凈的樣品溶液,確保進入高壓制備色譜系統的樣品無固體顆粒雜質,防止堵塞色譜柱,保證色譜系統正常運行,同時合適的溶劑選擇有助于樣品在色譜柱上的分離;選用反相c18柱,以乙腈-水為流動相,在特定的泵流速(0.8-1.2ml/min)、壓力(10-20mpa)和柱溫(30-40℃)條件下進行分離;反相c18柱對產物ⅰ和產物ⅱ具有不同的保留能力,通過調節乙腈-水的比例和色譜操作參數,使產物ⅰ和產物ⅱ根據其在固定相和流動相之間的分配差異實現精確分離,利用檢測器記錄色譜峰,根據出峰時間用餾分收集器收集含產物ⅰ和產物ⅱ的洗脫液餾分,得到高純度的產物ⅰ和產物ⅱ,且控制產物ⅰ和產物ⅱ的比例為1:0.73-0.75,滿足對該雜質合成的要求,為后續研究或質量控制等提供高質量的樣品;收集的餾分經旋轉蒸發儀減壓濃縮和真空干燥箱干燥,進一步去除殘留溶劑,得到高質量的白色固體產物ⅰ和產物ⅱ,確保產物的純度和穩定性,使其能夠滿足各種應用需求;產物ⅰ和產物ⅱ的合成反應式如下:
44、。
45、本發明中高壓制備分離步驟,通過精心配制乙腈-水流動相,確保其均勻性,為色譜分離提供合適的洗脫條件;以少量甲醇或流動相溶解粗產物,并采用先少量加入攪拌再超聲輔助溶解及微孔濾膜過濾的方式,制備純凈的樣品溶液,防止雜質堵塞色譜柱;精確進樣和設置優化的色譜儀參數,利用反相c18柱實現產物ⅰ和產物ⅱ基于吸附和洗脫動態平衡的精確分離,依據色譜峰收集餾分初步純化;再經旋轉蒸發濃縮和真空干燥,有效去除溶劑,獲得干燥的高質量產物ⅰ和產物ⅱ,確保產物的高純度、高穩定性,滿足對該雜質合成的嚴格要求,為后續研究或質量控制提供可靠的樣品。
46、作為優選,所述步驟s3產物ⅰ和產物ⅱ的合成中s35高壓制備分離步驟具體包括以下操作:
47、量取比例為90:10-60:40(v/v)的乙腈和水緩慢混合,攪拌使其充分混合均勻,得到配制好的流動相;少量甲醇或之前配制好的流動相作為溶劑溶解粗產物(產物ⅰ和產物ⅱ的混合物),開始時,先取少量粗產物,逐漸加入溶劑并攪拌,待粗產物基本溶解后,使用超聲輔助溶解,將盛有樣品溶液的容器放入超聲清洗器中超聲處理5-10分鐘,確保粗產物充分溶解,溶解后的樣品溶液用0.22-0.45μm的微孔濾膜進行過濾,將濾膜放置在過濾器上,將樣品溶液緩慢倒入過濾器,利用真空泵或手動擠壓的方式使溶液通過濾膜,收集過濾后的樣品溶液;將過濾好的樣品溶液注入高壓制備色譜儀中,使用微量注射器或進樣閥精確進樣,進樣量在10-100μl;對高壓制備色譜儀進行操作參數設置,泵流速設置為0.8-1.2ml/min,壓力設定在10-20mpa,柱溫控制在30-40℃;開啟色譜儀,使用配備的反相c18柱進行分離操作,在流動相的推動下,產物ⅰ和產物ⅱ在柱內與固定相發生吸附和洗脫的動態平衡,不同的保留時間使它們先后從柱中流出,根據檢測器記錄的色譜峰,觀察峰的出現時間和峰形,在接近產物ⅰ或產物ⅱ的預計保留時間時,開啟餾分收集器,將相應的餾分收集到不同的容器中,實現兩者的初步純化;將收集到的含有產物ⅰ和產物ⅱ的洗脫液餾分分別轉移至圓底燒瓶中,使用旋轉蒸發儀進行濃縮操作,將圓底燒瓶安裝在旋轉蒸發儀上,調節水浴溫度在30-40℃,旋轉蒸發儀的壓力設置在-0.08--0.1mpa,使產物從溶液中析出,形成白色固體;將濃縮得到的產物ⅰ和產物ⅱ白色固體轉移至真空干燥箱的托盤上,設置真空干燥箱的溫度為40-50℃,真空度保持在-0.08--0.1mpa,干燥時間為2-4小時;干燥結束后,關閉真空干燥箱,待壓力恢復至常壓后打開干燥箱,得到干燥后的高質量的產物ⅰ和產物ⅱ。
48、量取比例為90:10-60:40(v/v)的乙腈和水配制流動相,乙腈和水的不同比例會影響流動相的極性,通過調整極性改變產物ⅰ和產物ⅱ在反相c18柱上的保留行為,合適的極性能夠使產物ⅰ和產物ⅱ在柱內與固定相的吸附和洗脫能力產生差異,從而實現兩者的有效分離,不同極性的流動相可以提供不同的選擇性,有助于根據產物的化學性質差異找到最佳的分離條件,提高分離效果和純度;緩慢混合乙腈和水并攪拌均勻,是為了保證流動相的組成在整個體系中一致,不均勻的流動相導致色譜峰變形、分離效果變差等問題,均勻的流動相能夠使產物在色譜柱中受到穩定的洗脫力,保證分離過程的重復性和準確性,為后續精確分離產物ⅰ和產物ⅱ奠定基礎;用少量甲醇或之前配制好的流動相溶解粗產物,先取少量粗產物逐漸加入溶劑并攪拌,這種方式可以避免一次性加入過多溶劑導致粗產物結塊而難以溶解,待粗產物基本溶解后使用超聲輔助溶解,超聲利用高頻振動產生的能量加速粗產物在溶劑中的溶解過程,確保粗產物充分溶解形成均勻的溶液,使樣品能夠以分子狀態進入色譜柱進行分離,提高分離效果;溶解后的樣品溶液用0.22-0.45μm的微孔濾膜過濾,該孔徑的濾膜可以有效去除溶液中的微小顆粒雜質,如未溶解的固體顆粒、灰塵等,這些雜質進入高壓制備色譜儀會堵塞色譜柱的填料孔隙,增加柱壓,影響色譜柱的性能和壽命,甚至導致色譜柱損壞,過濾后的純凈樣品溶液能夠保證色譜系統正常運行,確保分離過程的順利進行,提高分析結果的準確性;將過濾好的樣品溶液注入高壓制備色譜儀,使用微量注射器或進樣閥精確進樣,進樣量在10-100μl范圍內,精確的進樣量確保每次進樣的樣品量一致,在定量分析和產物分離過程中,準確的進樣量有助于準確計算產物的含量和收率,同時保證不同批次實驗結果的可比性,提高實驗的可靠性和準確性;對高壓制備色譜儀進行操作參數設置,泵流速設置為0.8-1.2ml/min,合適的泵流速可以控制樣品在色譜柱中的停留時間,影響產物與固定相和流動相之間的相互作用,從而影響分離效果,壓力設定在10-20mpa,保證流動相能夠穩定地通過色譜柱,同時避免壓力過高對色譜柱造成損害,柱溫控制在30-40℃,柱溫會影響樣品的擴散速度和色譜柱的選擇性,合適的柱溫有助于優化產物ⅰ和產物ⅱ在柱內的分離效果,提高分離度和峰形質量,使兩者能夠更好地分離;在色譜分離過程中,根據檢測器記錄的色譜峰,在接近產物ⅰ或產物ⅱ的預計保留時間時開啟餾分收集器,將相應的餾分收集到不同容器中,實現產物ⅰ和產物ⅱ的初步純化,將它們從復雜的混合物中分離出來,避免相互污染,得到相對純凈的含有產物ⅰ和產物ⅱ的洗脫液餾分,為后續進一步處理提供高純度的原料;將收集到的洗脫液餾分轉移至圓底燒瓶,用旋轉蒸發儀在30-40℃水浴、-0.08--0.1mpa壓力下減壓濃縮,使溶劑快速蒸發,產物從溶液中析出形成白色固體,低溫減壓濃縮避免產物在濃縮過程中因高溫或長時間受熱而發生分解、氧化等化學反應,保證產物的質量;最后將濃縮得到的產物ⅰ和產物ⅱ白色固體在真空干燥箱中,在40-50℃、-0.08--0.1mpa真空度下干燥2-4小時,進一步去除殘留溶劑,得到干燥后的高質量的產物ⅰ和產物ⅱ,使其能夠滿足高純度要求。
49、綜上所述,本發明具有以下有益效果:
50、1、與現有技術相比,本發明采用了新的合成路線,在中間體ⅰ合成中用醋酸酐對黃體酮3位酮基進行保護,在中間體ⅱ合成中選擇硼氫化鈉作為還原劑和設置反應條件為冰浴、氮氣保護、分三批加入硼氫化鈉等,以及在產物ⅰ和產物ⅱ合成中使用氫氧化鉀水溶液脫保護等一系列操作,使得最終產物ⅰ和產物ⅱ的比例達到1:0.73-0.75,相比專利ep3275888a1(產物ⅰ和產物ⅱ比例為1:0.1)和專利cn113788871a(產物ⅰ和產物ⅱ比例約為1:0.4),產物ⅱ的收率有了顯著提高,在相同的原料投入下,本發明的黃體酮合成方法可以獲得更多的產物ⅱ,提高了原料的利用率,降低了合成成本,對于大規模生產該雜質具有重要的經濟意義;
51、2、相比于現有的黃體酮合成工藝,本發明由于產物ⅱ收率的提高,在生產相同數量的產物ⅱ時,所需的原料量相對減少,在現有技術中,為了獲得一定量的產物ⅱ,需要投入大量的黃體酮等原料,而本發明通過優化合成方法,提高了反應的選擇性和收率,從而減少了原料的浪費,降低了物料成本,新的合成路線中所使用的試劑和催化劑等相對較為常見和經濟,進一步降低了合成成本;
52、3、本發明在各個合成步驟中,通過合理設計反應條件和采用有效的分離純化手段(多次過濾、萃取、打漿、高壓制備色譜等),使中間體和產物能夠更高效地分離和純化,相比于傳統的分離純化方法,本發明減少了復雜的操作流程和多次重復的純化步驟,降低了能源消耗和人力成本,同時也減少了在分離純化過程中因操作繁瑣而導致的產物損失;同時,本發明合成的產物ⅰ和產物ⅱ具有較高的純度,通過高壓制備色譜等精確的分離純化技術,可以得到高質量的雜質對照品;
53、4、區別于現有技術中,該雜質由于構型翻轉,合成過程面臨的反應條件苛刻、副反應多等困難,本發明通過詳細研究和優化每一步反應條件,通過溫度控制(室溫、冰浴等)、反應時間(1-3小時、1-2小時等)、試劑用量(黃體酮、對甲苯磺酸、硼氫化鈉、氫氧化鉀水溶液等的用量范圍)以及反應操作方式(如氮氣置換方式、硼氫化鈉分三批加入等),成功克服合成困難,使反應能夠順利進行,提高了合成的成功率和穩定性;
54、5、傳統的原料藥雜質分離純化方法在處理該雜質時,存在選擇性差、分離效率低等問題,難以獲得高純度的產物,本發明綜合運用多種分離純化技術,根據中間體和產物的性質,選擇合適的溶劑(如甲醇、乙酸乙酯等)、干燥劑(如無水硫酸鈉)和分離手段(如過濾、萃取、色譜分離等),并優化操作條件(如萃取時的攪拌速度、濃縮時的溫度和壓力等),實現了中間體和產物的高效分離純化,解決了現有技術中分離純化效果不佳的問題,為獲得高質量的產物提供了保障。
- 還沒有人留言評論。精彩留言會獲得點贊!