一種桑枝皮納米纖維素晶須的制備方法與流程
本發明涉及一種納米纖維素的技術領域,特別涉及一種桑枝皮納米纖維素晶須的制備方法。
背景技術:
纖維素是一種廣泛存在于自然界中的可再生有機高分子,具有無毒、環境友好、易于改性和低密度等優點,纖維素獨特的性質使其具有巨大的潛在應用價值。納米纖維素晶須是由纖維素經酸水解得到的產物,呈棒狀,具有楊氏模量高、表面張力大等特性,具有獨特的機械性能,在醫藥材料、離子吸附與交換材料、生物功能材料等方面有廣泛的應用。
目前制備纖維素納米晶須的方法主要有酸水解法、生物酶法和物理機械法,其中生物酶法和物理機械法由于價格昂貴、需要使用特殊的設備等原因應用并不廣泛,酸水解法利用滲透進入的酸電離出氫離子,使纖維素分子鏈的糖苷鍵催化斷裂,破除無定形區使纖維素降解,得到纖維素納米晶須。
傳統的酸解法直接將木材和棉絮等纖維原料浸于酸中進行酸解,這種一步酸解的方法制備的纖維素納米晶須粒徑不均勻,長徑比難以控制。
技術實現要素:
有鑒于此,本發明目的在于提供一種桑枝皮納米纖維素晶須的制備方法,利用兩步酸解的方法制備桑枝皮納米纖維素晶須,得到的纖維素納米晶須長徑比大,形態均一,適用作靜電紡納米膜和復合材料的增強相。
為了實現上述發明目的,本發明提供以下技術方案:
本發明提供了一種桑枝皮納米纖維素晶須的制備方法,包括以下步驟:
將桑枝皮纖維在硝酸乙醇溶液中進行第一酸解,得到桑枝皮微晶纖維素;
將所述桑枝皮微晶纖維素在硫酸溶液中進行第二酸解,得到桑枝皮納米纖維素晶須。
優選的,所述硝酸乙醇溶液中硝酸和乙醇的體積比為1:3~5。
優選的,所述桑枝皮纖維與硝酸乙醇溶液的浴比為1:23~25。
優選的,所述第一酸解的溫度為90~100℃;所述第一酸解的時間為1~25h。
優選的,所述硫酸溶液的質量濃度為60~70%。
優選的,所述桑枝皮微晶纖維素的質量與硫酸溶液的體積比為1g:8~15ml。
優選的,所述第二酸解的溫度為50~70℃;所述第二酸解的時間為15~30min。
優選的,所述第二次酸解后還包括:將所述第二酸解得到的產物依次離心、透析、超聲波清洗和干燥。
優選的,所述透析的時間為60~72h。
優選的,所述干燥為冷凍干燥。
本發明提供了一種桑枝皮納米纖維素晶須的制備方法,包括以下步驟:將桑枝皮纖維在硝酸乙醇溶液中進行第一酸解,得到桑枝皮微晶纖維素;將所述桑枝皮微晶纖維素在硫酸溶液中進行第二酸解,得到桑枝皮納米纖維素晶須。桑枝皮資源豐富、成本廉價,本發明采用桑枝皮纖維為原料制備納米纖維素晶須,又提高了桑枝皮資源綜合利用價值;并且本發明利用兩步酸解的方法制備桑枝皮納米纖維素晶須,得到納米纖維素晶須長徑比大,形態均一,適用于作為復合材料的增強相。實驗結果表明,本發明制備的桑枝皮纖維素納米晶須長度為120~200nm,寬度為20~60nm,表現出較大的長徑比。
附圖說明
圖1為本發明實施例1制備的桑枝皮纖維素納米晶須的掃描電子顯微鏡照片;
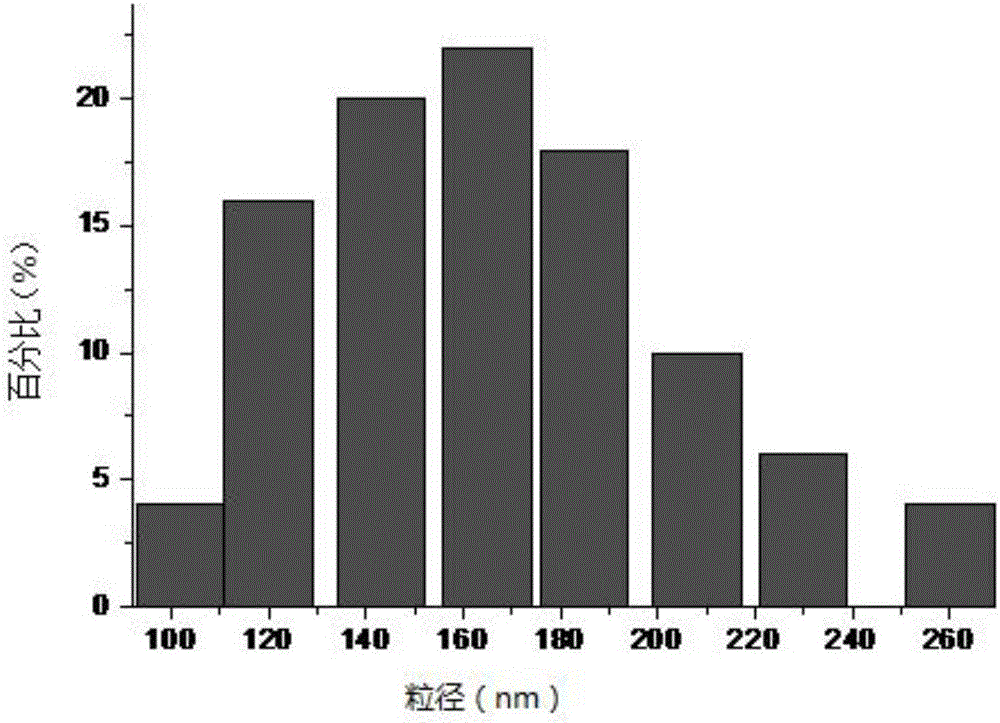
圖2為本發明實施例1制備的桑枝皮纖維素納米晶須的粒徑分布圖。
具體實施方式
本發明提供了一種桑枝皮納米纖維素晶須的制備方法,包括以下步驟:
將桑枝皮纖維在硝酸乙醇溶液中進行第一酸解,得到桑枝皮微晶纖維素;
將所述桑枝皮微晶纖維素在硫酸溶液中進行第二酸解,得到桑枝皮納米纖維素晶須。
本發明以桑枝皮纖維為原料制備納米纖維素晶須,桑樹枝條全桿分為木質部、韌皮部和髓芯,桑枝皮纖維來源于桑樹枝條的韌皮部分,約占全桿總質量的26~30%,桑枝皮纖維中含有多種不同的化學成分,主要包括纖維素部分和非纖維素部分,其中纖維素含量約為30%左右,非纖維素部分主要包括半纖維素、果膠、木質素、灰分等。
本發明對桑枝皮纖維的來源沒有特殊要求,可以使用機械方法從桑枝皮韌皮部分剝離得到,也可以直接以市售的桑枝皮纖維作為原料。
本發明將桑枝皮纖維在硝酸乙醇溶液中進行第一酸解,得到桑枝皮微晶纖維素。在本發明中,所述硝酸乙醇溶液由硝酸和乙醇配制得到,所述硝酸和乙醇的體積比優選為1:3~5,更優選為1:3.5~4.5,最優選為1:4;所述硝酸硝酸優選為質量分數為65%的濃硝酸;所述乙醇優選為無水乙醇。
本發明優選將硝酸加入乙醇中,得到硝酸乙醇溶液。本發明優選將硝酸分多份加入乙醇中,更優選將硝酸分為10~20份加入乙醇中,硝酸溶液的加入總體積符合上述比例;本發明優選在攪拌的條件下將硝酸溶液和乙醇混合。
在本發明中,所述桑枝皮纖維的長度優選為0.1~1cm,更優選為0.3~0.5cm。在本發明的部分具體實施例中,可以利用剪刀將桑枝皮纖維剪碎。
在本發明中,所述桑枝皮纖維與硝酸乙醇溶液的浴比優選為1:23~25,更優選為1:24。在本發明中,所述第一酸解的溫度優選為90~100℃,更優選為95~98℃;所述第一酸解的時間優選為1~25h,更優選為3~20h,最優選為5~15h;本發明優選在油浴的條件下進行加熱;本發明優選在冷凝回流的條件下進行第一酸解。
所述第一酸解后,發明優選觀察酸解液和酸解產物的顏色變化,若酸解液顏色發黑,酸解產物偏黃,則本發明優選重復第一酸解步驟,直至酸解產物顏色變白,第一酸解液沒有顏色變化為止;所述重復次數優選為1~5次,更優選為3~4次。
本發明利用硝酸乙醇溶液處理桑枝皮纖維,脫除桑枝皮中的非纖維素部分,使其中的木質素形成硝化木素或氧化木素溶解在乙醇中,同時大量的半纖維素及其他雜質被水解、氧化而溶出,從而達到將木質素和半纖維素等成分從桑枝皮纖維中脫除的目的;同時利用乙醇介質避免桑枝皮纖維中的纖維素被水解,從而得到純凈的桑枝皮微晶纖維素,進而使第二步酸解中無定形區的水解更加容易進行,使所得納米纖維素晶須的長徑比增大,粒徑更加均勻。
第一酸解后,本發明優選將得到的第一酸解液過濾,將過濾產物進行洗滌和干燥,得到桑枝皮微晶纖維素。本發明對過濾方法沒有特殊要求,使用本領域常規的過濾方法即可。
本發明優選使用硝酸乙醇溶液洗滌所述過濾產物1~2次,再用水洗滌產物至洗滌液pH為6.5~7.2,最后用無水乙醇洗滌1~3次。在本發明中,所述水洗滌中洗滌水的溫度優選為40~60℃,更優選為45~55℃。
在本發明中,所述干燥的溫度優選為90~110℃,更優選為95~105℃,所述干燥的時間優選為1~2h,更優選為1.5~1.8h。
所述干燥完成后,本發明將干燥產物進行粉碎,得到桑枝皮微晶纖維素。在本發明中,所述粉碎的粒度優選為20~80μm,更優選為30~50μm;本發明優選使用粉碎機進行粉碎。
得到桑枝皮微晶纖維素后,本發明將所述桑枝皮微晶纖維素在硫酸溶液中進行第二酸解,得到桑枝皮納米纖維素晶須。在本發明中,所述硫酸溶液的質量濃度優選為60~70%,更優選為63~68%,最優選為65%;所述桑枝皮微晶纖維素的質量與硫酸溶液的體積比優選為1g:8~15ml,更優選為1g:9~12ml。在本發明中,所述第二酸解的溫度優選為50~70℃,更優選為55~65℃;所述第二酸解的時間優選為15~30min,更優選為20~25min。
所述第二酸解后,本發明優選將得到的第二酸解液稀釋8~10倍使反應停止,得到桑枝皮納米纖維素晶須稀釋液;所述稀釋用稀釋劑優選為水,所述稀釋過程優選為將第二酸解液加入水中;所述水的體積為第二酸解液的7~9倍。
所述稀釋完成后,本發明優選將所述桑枝皮納米纖維素晶須稀釋液進行離心、透析和超聲處理,得到桑枝皮納米纖維素晶須懸浮液。在本發明中,所述離心優選在稀釋液靜置20~26h后進行,更優選為24h;所述離心的次數優選為3~6次,更優選為4~5次;所述單次離心的時間優選為15~25min,更優選為18~22min,最優選為20min;所述離心的轉速優選為3000~6000rpm,更優選為3500~5500rpm,最優選為4000rpm。
離心處理完成后,本發明將離心得到的懸浮液進行透析處理,所述透析處理的終點pH值優選為6.5~7.5,更優選為6.8~7.2,最優選為7.0;所述透析的時間優選為60~72小時,更優選為65~70h;所述透析用透析袋的截留分子量優選為8000~14000,更優選為10000~12000;本發明優選在透析過程中對透析用水進行更換,所述更換的頻率優選為10~15h/次,更優選為12h/次;本發明優選在透析開始時進行攪拌,以加速透析,所述攪拌優選為磁力攪拌,所述攪拌的時間優選為3~5h,更優選為4h。
所述透析完成后,本發明將透析得到的懸浮液進行超聲波清洗,得到桑枝皮納米纖維素晶須懸浮液。在本發明中,所述超聲波清洗的溫度優選為20~30℃,更優選為22~25℃;所述聲波清洗的時間優選為5~15min,更優選為8~12min,最優選為10min。
超聲清洗完成后,本發明優選將桑枝皮納米纖維素晶須懸浮液過濾,去除其中的不溶雜質;所述過濾優選使用砂芯漏斗。
得到桑枝皮納米纖維素晶須懸浮液后,本發明優選將桑枝皮納米纖維素晶須懸浮液干燥,得到桑枝皮納米纖維素晶須。在本發明中,所述干燥優選為冷凍干燥;所述冷凍干燥的溫度優選為-10~-30℃,更優選為-15~-25℃;所述冷凍干燥的時間優選為1~3h,更優選為1.5~2.5h。
下面結合實施例對本發明提供的桑枝皮納米纖維素晶須的制備方法進行詳細的說明,但是不能把它們理解為對本發明保護范圍的限定。
實施例1
稱取100g桑枝皮纖維,用剪刀將100g桑枝皮纖維剪碎,長度小于1cm;在400ml無水乙醇中緩慢加入100ml質量分數為65%的濃硝酸溶液,保證硝酸與乙醇的配比為1:4,攪拌均勻,每次加5ml,共加20次,備用;
將桑枝皮纖維與硝酸乙醇溶液混合,控制浴比為1:23,在90℃冷凝回流的條件下油浴加熱1h,充分攪拌,重復上述操作3~4次,直至濾液的顏色不變,纖維素顏色變白,將酸解液過濾,用硝酸乙醇繼續洗滌過濾產物2次,再用50℃熱水洗滌產物至中性,最后無水乙醇洗滌兩次,將洗滌后的產物在90℃下干燥1h,最后將干燥后的樣品粉碎至30μm左右得到桑枝皮纖維微晶纖維素;
取90ml質量濃度為65%的硫酸溶液,水浴加熱至55℃,向硫酸溶液中加入桑枝皮纖維微晶纖維素10g反應20min,到達反應時間后將溶液稀釋10倍使反應停止,將稀釋液靜置24h后進行離心,控制離心轉速為4000r/min,離心時間為20min,重復離心3次;
將離心得到的懸浮液進行透析處理,采用截留分子量為8000~14000的透析袋,把透析袋10cm的小段,用沸水將透析袋煮5分鐘,再用蒸餾水洗凈后使用,將要透析的桑枝皮纖維素納米晶須的液體倒入透析袋中進行透析,透析時間為72h,透析初期使用磁力攪拌器攪拌4小時加速透析,透析過程中每12小時換一次水,控制透析終點pH為7;
將透析得到的懸浮液在超聲波清洗器中清洗10min,水溫控制在25℃,使其分散均勻,用砂芯漏斗過濾去除不溶的雜質,得到分散均勻的桑枝皮纖維素納米晶須懸乳液;將桑枝皮纖維素納米晶須懸乳液在-10℃條件下冷凍干燥1h,得到桑枝皮纖維素納米晶須。
使用掃描電子顯微鏡對所得桑枝皮纖維素納米晶須進行形貌觀測,觀測結果如圖1所示;根據圖1可以看出,所得桑枝皮纖維素納米晶須粒徑均勻,為短棒狀結構,長度為幾百個納米,寬度在幾十個納米,表現出大的長徑比;
使用激光粒度儀對所得桑枝皮纖維素納米晶須的粒徑分布情況進行檢測,所得結果如圖2所示;根據圖2可以看出,80%的左右的桑枝皮納米纖維素晶須粒徑分布集中在120~200nm之間;
使用激光粒度儀對所得桑枝皮纖維素納米晶須的平均長度、平均直徑進行測定,并計算長徑比,將結果列于表1中。
實施例2
稱取100g桑枝皮纖維,用剪刀將100g桑枝皮纖維剪碎,長度約為0.5cm;在450ml無水乙醇中緩慢加入100ml質量分數為65%的濃硝酸溶液,保證硝酸與乙醇的配比為1:4.5,攪拌均勻,每次加5ml,共加20次,備用;
將桑枝皮纖維與硝酸乙醇溶液混合,控制浴比為1:24,在90℃冷凝回流的條件下油浴加熱25h,充分攪拌,重復上述操作3~4次,直至濾液的顏色不變,纖維素顏色變白,將酸解液過濾,用硝酸乙醇繼續洗滌過濾產物2次,再用50℃熱水洗滌產物至中性,最后無水乙醇洗滌兩次,將洗滌后的產物在100℃下干燥2h,最后將干燥后的樣品粉碎至50μm左右得到桑枝皮纖維微晶纖維素;
取100ml質量濃度為60%的硫酸溶液,水浴加熱至60℃,向硫酸溶液中加入桑枝皮纖維微晶纖維素10g反應15min,到達反應時間后將溶液稀釋10倍使反應停止,將稀釋液靜置24h后進行離心,控制離心轉速為4500r/min,離心時間為25min,重復離心3次;
將離心得到的懸浮液進行透析處理,采用截留分子量為8000~14000的透析袋,把透析袋10cm的小段,用沸水將透析袋煮5分鐘,再用蒸餾水洗凈后使用,將要透析的桑枝皮纖維素納米晶須的液體倒入透析袋中進行透析,透析時間為65h,透析初期使用磁力攪拌器攪拌4小時加速透析,透析過程中每12小時換一次水,控制透析終點pH為7;
將透析得到的懸浮液在超聲波清洗器中清洗15min,水溫控制在20℃,使其分散均勻,用砂芯漏斗過濾去除不溶的雜質,得到分散均勻的桑枝皮纖維素納米晶須懸乳液;將桑枝皮纖維素納米晶須懸乳液在-20℃條件下冷凍干燥1h,得到桑枝皮纖維素納米晶須。
使用掃描電子顯微鏡對所得桑枝皮纖維素納米晶須進行形貌觀測,觀測結果和實施例1相似;
使用激光粒度儀對所得桑枝皮纖維素納米晶須的粒徑分布情況進行檢測,所得結果和實施例1相似;
使用激光粒度儀對所得桑枝皮纖維素納米晶須的平均長度、平均直徑進行測定,并計算長徑比,將結果列于表1中。
實施例3
稱取100g桑枝皮纖維,用剪刀將100g桑枝皮纖維剪碎,長度小于1cm;在400ml無水乙醇中緩慢加入100ml質量分數為65%的濃硝酸溶液,每次加5ml,共加20次,保證硝酸與乙醇的配比為1:40,攪拌均勻,備用;
將桑枝皮纖維與硝酸乙醇溶液混合,控制浴比為1:25,在95℃冷凝回流的條件下油浴加熱5h,充分攪拌,重復上述操作3~4次,直至濾液的顏色不變,纖維素顏色變白,將酸解液過濾,用硝酸乙醇繼續洗滌過濾產物2次,再用45℃熱水洗滌產物至中性,最后無水乙醇洗滌兩次,將洗滌后的產物在90℃下干燥3h,最后將干燥后的樣品粉碎至25μm左右,得到桑枝皮纖維微晶纖維素;
取95ml質量濃度為63%的硫酸溶液,水浴加熱至65℃,向硫酸溶液中加入桑枝皮纖維微晶纖維素10g反應25min,到達反應時間后將溶液稀釋10倍使反應停止,將稀釋液靜置20h后進行離心,控制離心轉速為5000r/min,離心時間為20min,重復離心3次;
將離心得到的懸浮液進行透析處理,采用截留分子量為8000~14000的透析袋,將要透析的桑枝皮纖維素納米晶須的液體倒入透析袋中進行透析,透析時間為72h,透析初期使用磁力攪拌器攪拌4小時加速透析,透析過程中每12小時換一次水,控制透析終點pH為7;
將透析得到的懸浮液在超聲波清洗器中清洗12min,水溫控制在30℃,使其分散均勻,用砂芯漏斗過濾去除不溶的雜質,得到分散均勻的桑枝皮纖維素納米晶須懸乳液;將桑枝皮纖維素納米晶須懸乳液在-30℃條件下冷凍干燥2h,得到桑枝皮纖維素納米晶須。
使用掃描電子顯微鏡對所得桑枝皮纖維素納米晶須進行形貌觀測,觀測結果和實施例1相似;
使用激光粒度儀對所得桑枝皮纖維素納米晶須的粒徑分布情況進行檢測,所得結果和實施例1相似;
使用激光粒度儀對所得桑枝皮纖維素納米晶須的平均長度、平均直徑進行測定,并計算長徑比,將結果列于表1中。
實施例4
用剪刀將桑枝皮纖維剪碎,長度小于1cm;在500ml無水乙醇中緩慢加入100ml質量分數為65%的濃硝酸溶液,每次加5ml,共加20次,保證硝酸與乙醇的配比為1:40,攪拌均勻,備用;
將桑枝皮纖維與硝酸乙醇溶液混合,控制浴比為1:25,在100℃冷凝回流的條件下油浴加熱20h,充分攪拌,重復上述操作3~4次,直至濾液的顏色不變,纖維素顏色變白,將酸解液過濾,用硝酸乙醇繼續洗滌過濾產物2次,再用45℃熱水洗滌產物至中性,最后無水乙醇洗滌兩次,將洗滌后的產物在95℃下干燥2h,最后將干燥后的樣品粉碎至60μm左右,得到桑枝皮纖維微晶纖維素;
取100ml質量濃度為70%的硫酸溶液,水浴加熱至50℃,向硫酸溶液中加入桑枝皮纖維微晶纖維素10g反應30min,到達反應時間后將溶液稀釋10倍使反應停止,將稀釋液靜置25h后進行離心,控制離心轉速為5500r/min,離心時間為20min,重復離心3次;
將離心得到的懸浮液進行透析處理,采用截留分子量為8000~14000的透析袋,將要透析的桑枝皮纖維素納米晶須的液體倒入透析袋中進行透析,透析時間為60h,透析初期使用磁力攪拌器攪拌4小時加速透析,透析過程中每12小時換一次水,控制透析終點pH為7;
將透析得到的懸浮液在超聲波清洗器中清洗5min,水溫控制在10℃,使其分散均勻,用砂芯漏斗過濾去除不溶的雜質,得到分散均勻的桑枝皮纖維素納米晶須懸乳液;將桑枝皮纖維素納米晶須懸乳液在-25℃條件下冷凍干燥2h,得到桑枝皮纖維素納米晶須。
使用掃描電子顯微鏡對所得桑枝皮纖維素納米晶須進行形貌觀測,觀測結果和實施例1相似;
使用激光粒度儀對所得桑枝皮纖維素納米晶須的粒徑分布情況進行檢測,所得結果和實施例1相似;
使用激光粒度儀對所得桑枝皮纖維素納米晶須的平均長度、平均直徑進行測定,并計算長徑比,將結果列于表1中。
表1實施例1~4所得桑枝皮纖維素納米晶須長徑比分析
根據表1可以看出,本發明提供的制備方法得到的桑枝皮纖維素納米晶須粒徑均勻,且粒徑分布范圍窄,長徑比可以達到4.33。
由以上實施例可知,本發明由所述的兩步酸解制備桑枝皮纖維素納米晶須的方法簡單易控,且所得的桑枝皮纖維素納米晶須尺寸均勻,長徑比大,適用于作為復合材料的增強相。
以上所述僅是本發明的優選實施方式,應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!