一種高效的纖維素納米晶制備方法與流程
【技術領域】
本發明屬于納米材料制備技術領域,特別涉及一種纖維素納米晶的綠色、環保的宏量制備方法。
背景技術:
纖維素是自然界中存在量最大的天然生物材料,廣泛存在于綠色植物、海洋動物等生物體中。通過除去纖維素材料中的非晶區,可以得到納米尺寸的纖維素納米晶。纖維素納米晶具有低密度、高強度、高長徑比等特點;此外,纖維素納米晶還具有高比表面積、可反應表面、結晶度高等優點,是一種性能優良的增強劑,在高性能復合材料制備領域具有巨大的應用價值。
現有技術中,纖維素納米晶的制備主要采用酸解法或tempo試劑氧化法。酸解法制備纖維素納米晶大多采用硫酸、鹽酸、磷酸、硝酸等強酸或幾種酸的混合酸,利用其氧化性除去纖維素材料中的非晶區制備纖維素納米晶,如專利號為cn101509209b、cn1171904c、cn1142185c和cn102040663b的中國發明專利均采用酸解法制備纖維素納米晶,纖維素與強酸的用量質量比為1:14~1:60,現有酸解法制備纖維素納米晶的技術不僅存在因耗酸量大會對環境造成嚴重污染的問題,而且硫酸酸解法制備的纖維素納米晶表面帶有磺酸根,導致纖維素納米晶耐熱性差。此外,現有制備纖維素納米晶的技術還存在產率低(不超過60%)的缺點,故酸解法不適合宏量制備纖維素納米晶。tempo氧化法制備表面帶有羧基的纖維素納米晶需要用到tempo、溴化鈉、次氯酸鈉等多種化學試劑,而tempo試劑價格昂貴,此外,tempo氧化法制備纖維素納米晶的過程復雜,羧基含量嚴格依賴反應體系的ph條件,工藝條件苛刻,亦不適合纖維素納米晶的宏量制備。因此,開發一種綠色、環保的纖維素納米晶的宏量制備方法具有重要意義。
技術實現要素:
[要解決的技術問題]
本發明的目的在于提供一種綠色、環保的纖維素納米晶的宏量制備方法。
本發明的另一目的還在于提供一種通過控制金屬氧酸鹽和有機酸加入量的方法調控所制纖維素納米晶的長度,提高纖維素納米晶產率的方法。
本發明的另一目的還在于提供一種通過控制金屬氧酸鹽和有機酸加入量調控所制纖維素納米晶表面的羧基含量的方法。
[技術方案]
本發明的技術方案是針對目前纖維素納米晶制備成本高,且易造成環境污染等問題,提出的一種綠色環保的制備纖維素納米晶的方法。該方法利用金屬氧酸鹽在酸性環境中具有強氧化性,在反應體系中加入有機酸,不但為金屬氧酸鹽反應體系提供酸性環境,并且有機酸與含氧酸鹽之間的特殊相互作用可使金屬氧酸鹽的氧化性進一步提高,將原料纖維素的非晶區水解除去,得到能夠在水中穩定分散的表面帶有羧基的纖維素納米晶。
一種綠色、環保的纖維素納米晶的宏量制備方法,其特征在于主要包括以下步驟:
(1)預處理:將纖維素原料攪拌粉碎,用堿液浸泡1-120h,優選5-80h,更優選8-30h,然后用去離子水抽濾洗滌至中性;
(2)將步驟(1)所得處理后的纖維素分散在質量百分濃度為0.5%-40%,優選5%-30%,更優選10%-25%的金屬氧酸鹽水溶液中,加入有機酸,加熱攪拌。
(3)后處理:將步驟(2)所得水解液進行離心分散,取上層懸浮液透析、超聲分散,得到穩定分散的纖維素納米晶懸浮液。
所述步驟(1)中纖維素原料為微晶纖維素、植物纖維素、紙漿纖維素或α-纖維素;也可以是自然界中常見的天然纖維素,對于纖維素的種類在本發明中沒有限制。
所述步驟(1)中用堿液浸泡預處理時,所用堿溶液為質量百分含量為1%-10%,優選3%-8%,更優選4%-6%的氫氧化鋰、氫氧化鈉、氫氧化鉀、強氧化鈣、碳酸氫鈉、氫氧化鎂、氫氧化鐵、氫氧化亞鐵或氨溶液。
所述步驟(1)中纖維素浸泡于堿溶液中,纖維素原料與堿溶液的用量比為10-100g:1l,所述浸泡時間為1-120h。
所述步驟(2)中纖維素原料與金屬氧酸鹽的質量比為1:0.2~1:9。
所述步驟(2)中所用金屬氧酸鹽為高鐵酸鉀、高鐵酸鈉、硝酸鉀、硝酸鈉、氯酸鉀、氯酸鈉、次氯酸鉀、次氯酸鈉、高氯酸鉀、高氯酸鈉、溴酸鉀、溴酸鈉、次溴酸鉀、次溴酸鈉、高溴酸鉀、高溴酸鈉、鉍酸鉀或鉍酸鈉,優選高鐵酸鉀、高鐵酸鈉、氯酸鉀、氯酸鈉、鉍酸鉀、鉍酸鈉中的一種或幾種。本發明所用金屬氧酸鹽的范圍不僅限于此。
所述步驟(2)中反應溫度為50-150℃,反應時間為5-72h。
所述步驟(2)中所用酸為包含甲酸、乙酸、丙酸、丁酸、蘋果酸、檸檬酸或琥珀酸的有機酸,優選蘋果酸、檸檬酸和琥珀酸中的一種或幾種。
所述步驟(2)中金屬氧酸鹽和有機酸的質量比為1:2~1:0.1。
所述步驟(3)的水解液進行離心分離時,離心機轉速為3000~12000rpm,離心次數2-8次。
所述步驟(3)的水解液進行透析時,將離心分離后的溶液轉移到透析袋中透析3天至ph成中性。
所述制備方法得到的纖維素納米晶產率超過70%,纖維素納米晶表面羧基含量為0.5mmol/g~50mmol/g,長度為195~260nm。
[有益效果]
本發明由于采取上述技術方案,具有以下優點:
1.所用金屬氧酸鹽氧化劑和有機酸均是綠色、環保的試劑,對環境污染性小,且均為工業化產品,價格低廉。
2.所用金屬氧酸鹽在酸性環境中具有超強的氧化性,在反應體系中加入有機酸可以大大提高金屬氧酸鹽的氧化性,在制備纖維素納米晶過程中其用量較少,其用量與現有酸解法制備纖維素納米晶技術相比,減少量可達80%,在降低了制備成本的同時減小了對環境的污染,可用于宏量制備纖維素納米晶。
3.所制備的纖維素納米晶表面帶有羧基,羧基的存在一方面克服了纖維素納米晶因氫鍵易團聚的問題,使得所制備的纖維素納米晶容易均勻分散到水、n,n-二甲基甲酰胺、冰醋酸等體系中,拓展了纖維素納米晶的應用范圍;另一方面可以利用羧基與氨基和羥基等基團之間的可反應性,豐富了纖維素納米晶的改性途徑。
4.通過調控反應體系中金屬氧酸鹽和有機酸的加入量可調控所得cnc的長度及產率,纖維素納米晶產率可超過70%。
5.與硫酸酸解法所制cnc相比,本發明所制cnc耐熱性提高了50~100℃。
【附圖說明】
圖1是本發明實施例1所制備的纖維素納米晶的原子力顯微鏡照片。
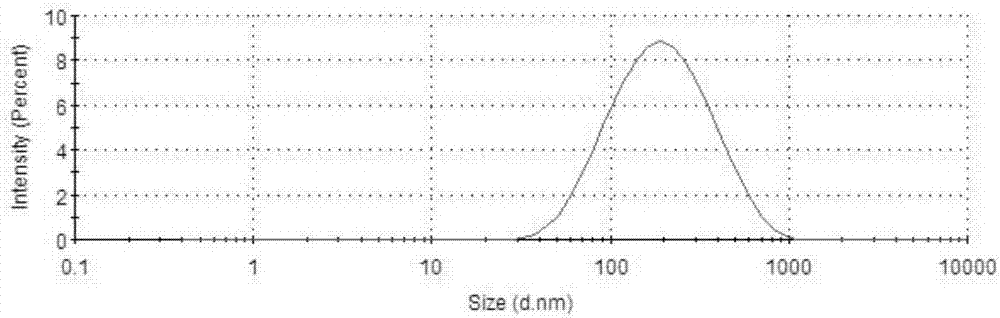
圖2是本發明實施例1所制備的纖維素納米晶的粒徑分布曲線。
圖3是本發明實施例1所制備的纖維素納米晶的紅外譜圖。
【具體實施方式】
下面結合實施例對本發明進一步描述,但本發明的保護范圍并不僅限于此。
實施例1
將5g紙漿纖維素用攪拌機打碎成棉絮狀,加入250ml濃度為4%的氫氧化鈉溶液,浸泡24h后,將其抽濾、洗滌至中性,再將堿預處理的纖維素分散于300ml去離子水中,加入2.5g高鐵酸鉀和1ml蘋果酸,90℃攪拌6h后,用高速離心機以8000rpm/min的轉速將纖維素水解液離心10min,然后將沉淀洗滌,再次分離,直至上清液變渾濁。將所得纖維素納米晶懸浮液超聲,得到穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶平均長度約為260nm,產率為60%,粒徑分散指數(pdi)為0.3,羧基含量為0.5mmol/g。
所制備的纖維素納米晶的形貌、粒徑分布如圖1和圖2,所制備的表面含有羧基的纖維素納米晶的紅外譜圖如圖3。從圖1中可以看出這種方法制備的纖維素納米晶呈典型的棒狀結構,且呈單分散狀態,說明纖維素納米晶之間沒有發生聚集;從圖2中可以看出所制備的纖維素納米晶粒徑分布指數較窄,說明所制備的纖維素納米晶長度分布比較均勻;從圖3中看出,通過對比本發明所述方法制備的纖維素納米晶和原料纖維素的紅外譜圖,從中可以看出,與原料纖維素相比,纖維素納米晶的紅外譜圖新增了1733cm-1處的羧基特征吸收峰,說明本申請的方法可制備表面帶有羧基的纖維素納米晶。
實施例2
所用原料種類、用量及工藝流程同實施例1,不同的是所用高鐵酸鉀的量為5g,得穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為220nm,產率為69%,pdi為0.2,羧基含量為6.5mmol/g。
實施例3
所用原料種類、用量及工藝流程同實施例1,不同的是所用高鐵酸鉀的量為7.5g,得穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為210nm,產率為75%,pdi為0.2,羧基含量為9.2mmol/g。
實施例4
所用原料種類、用量及工藝流程同實施例1,不同的是所用蘋果酸的量為2ml,得穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為245nm,產率為70%,pdi為0.2,羧基含量為13.6mmol/g。
實施例5
所用原料種類、用量及工藝流程同實施例1,不同的是所用蘋果酸的量為3ml,得穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為238nm,產率為71%,pdi為0.2,羧基含量為26.2mmol/g。
實施例6
所用原料種類、用量及工藝流程同實施例1,不同的是所用高鐵酸鉀的量為5g,蘋果酸的量為2ml,得穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為200nm,產率為73%,pdi為0.2,羧基含量為35.2mmol/g。
實施例7
所用原料種類、用量及工藝流程同實施例1,不同的是所用高鐵酸鉀的量為5g,蘋果酸的量為3ml,得穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為195nm,產率為76%,pdi為0.2,羧基含量為50.0mmol/g。
實施例8
所用原料種類、用量及工藝流程同實施例1,不同的是將紙漿纖維素在堿溶液中的浸泡時間延長至72h,得穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為220nm,產率為69%,pdi為0.2,羧基含量為12.9mmol/g。
實施例9
所用原料種類、用量配比及工藝流程同實施例1,不同的是制備纖維素納米晶時反應溫度為100℃,制得穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為225nm,產率為71%,pdi為0.3,羧基含量為14.1mmol/g。
實施例10
所用原材料種類、用量配比及工藝流程同實施例1,不同的是制備纖維素納米晶時反應時間為24h,得穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為215nm,產率為75%,pdi為0.2,羧基含量為40.1mmol/g。
實施例11
所用原料種類及工藝流程同實施例1,不同的是將紙漿纖維素的質量增加到100g,高鐵酸鉀和蘋果酸的用量也相應成比例增加,在90℃下攪拌6h。反應后處理亦同實施例1,經測量所得纖維素納米晶粒徑平均長度約為280nm,產率為72%,pdi為0.4,羧基含量為20.1mmol/g。
實施例12
所用原料種類及工藝流程同實施例1,不同的是將紙漿纖維素的質量增加到500g,高鐵酸鉀和蘋果酸的用量也相應成比例增加,在90℃下攪拌6h。反應后處理亦同實施例1,經測量所得纖維素納米晶粒徑平均長度約為285nm,產率為71%,pdi為0.3,羧基含量為19.5mmol/g。
實施例13
所用原料種類及工藝流程同實施例1,不同的是將紙漿纖維素的質量增加到1000g,高鐵酸鉀和蘋果酸的用量也相應成比例增加,在90℃下攪拌6h。反應后處理亦同實施例1,經測量所得纖維素納米晶粒徑平均長度約為290nm,產率為73%,pdi為0.5,羧基含量為25.8mmol/g。
實施例14
所用原料種類及工藝流程同實施例1,不同的是所用金屬氧酸鹽為鉍酸鈉,得到穩定的纖維素納米晶懸浮液,經測量所得纖維素納米晶粒徑平均長度約為195nm,產率為72%,pdi為0.3,羧基含量為24.2mmol/g。
對比例1
所用原料種類及工藝流程同實施例1,不同的是不加入有機酸,經氧化水解所得纖維素微纖的長度為1.5μm,遠大于實施例1中所得納米晶長度,纖維素微纖的產率為28%,遠低于實施例1中的產率,且其長度分布也較寬,pdi為0.89,羧基含量為0.1mmol/g,這說明有機酸的加入對提高金屬氧酸鹽的氧化性具有很重要的作用
對比例2
所用原料種類及工藝流程同實施例1,不同的是加入堿性調節劑劑氨水,使反應體系呈堿性環境,經氧化水解所得纖維素微纖的長度為5.1μm,遠大于實施例1中所得納米晶長度,纖維素微纖的產率為8%,遠低于實施例1中的產率,且其長度分布也較寬,pdi為1.00,羧基含量為0.09mmol/g,這說明堿性調節劑不但不能提高金屬氧酸鹽的氧化性,還對金屬氧酸鹽本身具有的弱氧化性有抑制作用。
對比例3
所用材料種類及工藝流程同實施例14,不同的是不加入有機酸,經氧化水解所得纖維素微纖的長度為1.2μm,遠大于實施例14中所得納米晶長度,纖維素微纖的產率為30%,遠低于實施例14中的產率,且其長度分布也較寬,pdi為0.79,羧基含量為0.05mmol/g。
對比例4
所用材料種類及工藝流程同實施例14,不同的是加入堿性調節劑氨水,使反應體系呈堿性環境,經氧化水解所得纖維素微纖的長度為4.2μm,遠大于實施例14中所得納米晶長度,纖維素微纖的產率為13%,遠低于實施例14中的產率,且其長度分布也較寬,pdi為0.98,羧基含量為0.05mmol/g。
對比例5
所用原料種類及工藝流程同實施例1,不同的是不進行堿預處理,經氧化水解所得纖維素納米晶的長度為750nm,大于實施例1中所得納米晶長度,纖維素納米晶的產率為36%,遠低于實施例1中的產率,pdi為0.67,羧基含量為0.01mmol/g,這說明用堿對纖維素進行預處理可以使纖維素的微纖間發生溶脹,有助于水解液進入纖維素內部從而促進水解反應進行。
對比例6
所用原料種類及工藝流程同實施例1,不同的是,高鐵酸鉀和有機酸的加入量分別為2.5g和6ml,經氧化水解所得纖維素微纖的長度為15nm,遠小于實施例1中所得納米晶長度,纖維素微纖的產率為5%,遠低于實施例1中的產率,且其長度分布也較寬,pdi為1,羧基含量為50mmol/g,這說明金屬氧酸鹽和有機酸的用量比小于0.5時,不利于水解反應可控進行。
對比例7
所用原料種類及工藝流程同實施例1,不同的是,高鐵酸鉀和有機酸的加入量分別為2.5g和0.15ml,經氧化水解所得纖維素微纖的長度為1um,遠大于實施例1中所得納米晶長度,纖維素微纖的產率為12%,遠低于實施例1中的產率,且其長度分布也較寬,pdi為1,羧基含量為0.09mmol/g,這說明金屬氧酸鹽和有機酸的用量比大于10時,有機酸的存在對金屬氧酸鹽的氧化性提高作用小,不利于水解反應進行。
表1
- 還沒有人留言評論。精彩留言會獲得點贊!