一種1-BOC-2,2-二甲基哌啶-4-酮的制備方法與流程
本發明涉及藥物化學合成,尤其涉及一種1-boc-2,2-二甲基哌啶-4-酮的制備方法。
背景技術:
1、1-boc-2,2-二甲基哌啶-4-酮是一種應用廣泛的醫藥中間體,尤其在新藥物的研究中起到重要作用。如公開號為us2004/0006229?a1的美國專利公開文本中記載,1-boc-2,2-二甲基哌啶-4-酮可作為中間體制備5-羥色胺(5-ht)激動劑,用于抑制神經原蛋白質外滲和治療或預防偏頭痛;又如公開號為wo?2016/046755的國際專利公開文本中記載,1-boc-2,2-二甲基哌啶-4-酮可作為中間體制備調節roryt活性的化合物,用于治療免疫和發炎性障礙疾病等方面的疾病。
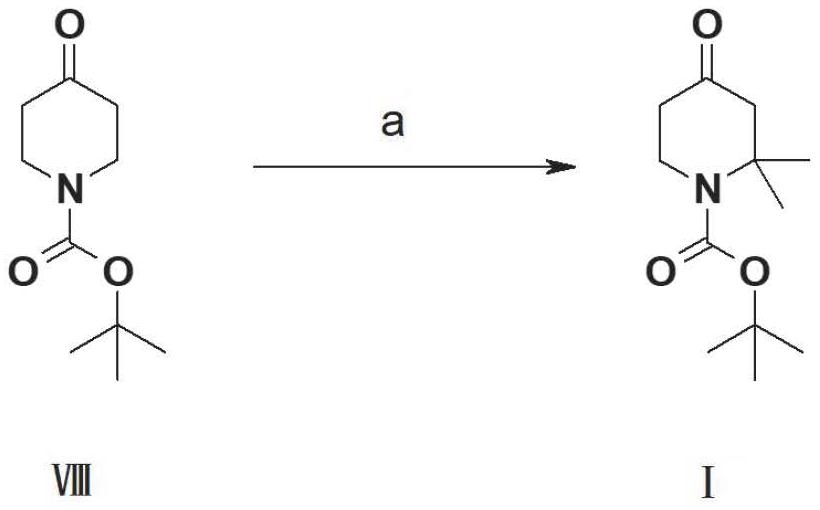
2、關于1-boc-2,2-二甲基哌啶-4-酮的合成,專利us2005/239781中報道了如下的制備方法:
3、
4、試劑與收率:(a)氫化鈉,碘甲烷,四氫呋喃,0~20℃°,12h,32%。
5、該方法以化合物ⅷ為原料,使用危險試劑氫化鈉,有異構副產物,收率低,難于純化,因此不適宜大規模生產。
6、關于1-boc-2,2-二甲基哌啶-4-酮的合成,專利us2003/232833報道了另外一種類似的制備方法如下:
7、
8、試劑:(a)叔丁酸鉀,四氫呋喃,0~20℃,4.5h;(b)乙酸乙酯,5n?hcl,回流,14.5h;(c)氫氧化鈉,二碳酸二叔丁酯,四氫呋喃,20℃,96h,ph=8~9。
9、該方法以化合物ix為原料,先經過關環、脫羧基-脫叔丁氧羰基保護基、上叔丁氧羰基。反應過程中原料昂貴不易得,用柱層析純化,不適宜大規模制備。專利中介紹了1-boc-2,2-二甲基哌啶-4-酮,以化合物?為原料采用上述類似方法制備得到,同樣原料昂貴不易得,且無收率報道,需要嘗試。其制備方法大致如下:
10、
技術實現思路
1、針對現有技術中對于1-boc-2,2-二甲基哌啶-4-酮的路線和制備方法原料昂貴不易得,產率低,分離純化困難等問題,本發明提供了1-boc-2,2-二甲基哌啶-4-酮的制備方法,反應式如下所示:
2、
3、包括以下步驟:
4、s1、將式ⅱ化合物溶于溶劑中,加入芐胺,加料完成后,升溫到60-65度,然后攪拌32小時,減壓濃縮至干,得到無色液體,加入甲基叔丁基醚,向混合物中加入10%的檸檬酸溶液,兩層分離,分出有機層,有機層無水硫酸鎂干燥,減壓濃縮,得到無色液體,即式ⅲ化合物;
5、s2、將式ⅲ化合物溶于四氫呋喃中,氮氣保護,加入堿1,控溫在10℃以下,滴加氯甲酰乙酸甲酯,將反應混合物自然升溫至室溫20℃,然后攪拌12小時,加入10%碳酸氫鈉水溶液,分出有機層,有機層用10%氯化鈉水溶液洗滌,分液,無水硫酸鎂干燥,過濾,減壓濃縮,得到棕色固體,加正庚烷/乙酸乙酯打漿,抽濾,濾餅得到黃色固體,即式ⅳ化合物;
6、s3、將式ⅳ化合物溶于甲醇中,氮氣保護,控制溫度在10度左右,分批緩慢加入堿2,加料完畢,加熱升溫到65-70度,攪拌2h,減壓濃縮掉甲醇,加入到2n鹽酸中,加入甲基叔丁基醚,分出有幾層,水相用甲基叔丁基醚萃取,合并有機相,無水硫酸鎂干燥,過濾,減壓濃縮得黃色固體,即式ⅴ化合物;
7、s4、將式ⅴ化合物溶于水和乙酸混合溶液中,加熱升溫到90-100度,控攪拌12小時,降溫到20度,緩慢滴加飽和碳酸鈉水溶液,攪拌析出大量固體,抽濾,濾餅用水漂洗,過濾,固體懸浮于正庚烷和甲基叔丁基醚的混合溶液中,攪拌打漿,得到黃色固體,即式ⅵ化合物;
8、s5、將式ⅵ化合物溶于四氫呋喃中,氮氣保護,控溫10度左右,分批加入硼氫化鈉,加料完畢,緩慢滴加三氟化硼乙醚,加料過程,放熱放氣,加完后升溫到55度,攪拌12小時,降溫到10度,緩慢滴加甲醇,放熱放氣,滴加完畢,升溫回流,攪拌6h,減壓濃縮反應液,加入2n鹽酸水溶液,攪拌30分鐘,向體系加入甲基叔丁基醚萃取,重復該操作兩次,合并有機相,無水硫酸鎂干燥,過濾,減壓濃縮得到類白色固體,即式ⅶ化合物;
9、s6、將式ⅶ化合物溶于甲醇中,加入(boc)2o和10%鈀碳,加畢高壓釜通入氫氣,在50℃下攪拌反應16h,體系抽濾,濾液濃縮,除去溶劑,加入正庚烷打漿,得到白色固體物,即式ⅰ化合物。
10、優選地,s1中,溶劑選自甲醇、乙醇或水的一種或兩種的混合物。
11、優選地,s2中,式ⅲ化合物:堿1:氯甲酰乙酸甲酯的摩爾比為1:1~1.5:1~2:1。
12、優選地,s2中,堿1為三乙胺、n,n'-二異丙基乙胺或1,8-二氮雜雙環[5.4.0]十一碳-7-烯。
13、優選地,s3中,堿2為叔丁醇鉀、叔丁醇鈉、甲醇鈉、乙醇鈉或者氫化鈉。
14、優選地,式ⅴ至式ⅵ化合物的反應中,酸為三氟乙酸、鹽酸、硫酸、乙酸或對甲苯磺酸。
15、優選地,式ⅵ至式ⅶ化合物的反應中,還原劑選自氫化鋰鋁、硼氫化鈉、三氟化硼乙醚、硼烷四氫呋喃絡合物、硼烷二甲硫醚絡合物或紅鋁。
16、優選地,式ⅵ至式ⅶ化合物的反應中,式ⅵ化合物與還原劑的摩爾比為1:1~1:1.5。
17、優選地,式ⅶ至式ⅰ化合物的反應中,催化劑選自pd/c,pd(oh)2/c或雷尼鎳。
18、更優選地,式ⅶ至式ⅰ化合物的反應中,氫氣壓力為2~4個大氣壓。
19、本發明的有益效果是:提供了一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,以3,3-二甲基丙烯酸甲酯、芐胺作為起始原料,經加成、酰化、dieckmann縮合,脫羧、還原、加氫等步驟制得,原料廉價易得且產率較高,操作簡便,可控性好,總體收率合適,適宜大規模生產。
技術特征:
1.一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:反應式如下所示:
2.根據權利要求1所述的一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:s1中,溶劑選自甲醇、乙醇或水的一種或兩種的混合物。
3.根據權利要求1所述的一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:s2中,式ⅲ化合物:堿1:氯甲酰乙酸甲酯的摩爾比為1:1~1.5:1~2:1。
4.根據權利要求3所述的一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:s2中,堿1為三乙胺、n,n'-二異丙基乙胺或1,8-二氮雜雙環[5.4.0]十一碳-7-烯。
5.根據權利要求1所述的一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:s3中,堿2為叔丁醇鉀、叔丁醇鈉、甲醇鈉、乙醇鈉或者氫化鈉。
6.根據權利要求1所述的一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:式ⅴ至式ⅵ化合物的反應中,酸為三氟乙酸、鹽酸、硫酸、乙酸或對甲苯磺酸。
7.根據權利要求1所述的一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:式ⅵ至式ⅶ化合物的反應中,還原劑選自氫化鋰鋁、硼氫化鈉、三氟化硼乙醚、硼烷四氫呋喃絡合物、硼烷二甲硫醚絡合物或紅鋁。
8.根據權利要求1所述的一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:式ⅵ至式ⅶ化合物的反應中,式ⅵ化合物與還原劑的摩爾比為1:1~1:1.5。
9.根據權利要求1所述的一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:式ⅶ至式ⅰ化合物的反應中,催化劑選自pd/c,pd(oh)2/c或雷尼鎳。
10.根據權利要求1所述的一種1-boc-2,2-二甲基哌啶-4-酮的制備方法,其特征在于:式ⅶ至式ⅰ化合物的反應中,氫氣壓力為2~4個大氣壓。
技術總結
本發明屬于涉及藥物化學合成技術領域,尤其涉及一種1?BOC?2,2?二甲基哌啶?4?酮的制備方法,包括Ⅲ化合物的制備,式Ⅳ化合物的制備,式Ⅴ化合物的制備,式Ⅵ化合物的制備,式Ⅶ化合物的制備以及式Ⅰ化合物的制備。本發明的有益效果是,原料易得、價格便宜,操作方便,反應易于控制,無副產物,總體收率合適,適宜大規模生產。
技術研發人員:徐明,孫光明,黃爾青,劉冬平
受保護的技術使用者:南京艾康生物科技有限公司
技術研發日:
技術公布日:2025/4/24
- 還沒有人留言評論。精彩留言會獲得點贊!